 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modifying the properties of 4f single-ion magnets by peripheral ligand functionalisation†
Kasper S.
Pedersen
a,
Liviu
Ungur
b,
Marc
Sigrist
ac,
Alexander
Sundt
d,
Magnus
Schau-Magnussen
a,
Veacheslav
Vieru
b,
Hannu
Mutka
c,
Stephane
Rols
c,
Høgni
Weihe
a,
Oliver
Waldmann
d,
Liviu F.
Chibotaru
b,
Jesper
Bendix
*a and
Jan
Dreiser
*e
aDepartment of Chemistry, University of Copenhagen, DK-2100 Copenhagen, Denmark. E-mail: bendix@kiku.dk
bTheory of Nanomaterials Group, Katholieke Universiteit Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
cInstitut Laue-Langevin, 38042 Grenoble Cedex 9, France
dPhysikalisches Institut, Universität Freiburg, 79104 Freiburg, Germany
eEcole Polytechnique Fédérale de Lausanne, Institute of Condensed Matter Physics, 1015 Lausanne and Swiss Light Source, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland. E-mail: jan.dreiser@epfl.ch
First published on 6th January 2014
Abstract
We study the ligand-field splittings and magnetic properties of three ErIII single-ion magnets which differ in the peripheral ligand sphere but exhibit similar first coordination spheres by inelastic neutron scattering (INS) and SQUID magnetometry. The INS spectra of the three compounds are profoundly different pointing at a strong response of the magnetic behavior to minor structural changes, as they are e.g. encountered when depositing molecules on surfaces. The observation of several magnetic excitations within the J = 15/2 ground multiplet together with single-crystal magnetic measurements allows for the extraction of the sign and magnitude of all symmetry-allowed Stevens parameters. The parameter values and the energy spectrum derived from INS are compared to the results of state-of-the-art ab initio CASSCF calculations. Temperature-dependent alternating current (ac) susceptibility measurements suggest that the magnetisation relaxation in the investigated temperature range of 1.9 K < T < 5 K is dominated by quantum tunnelling of magnetisation and two-phonon Raman processes. The possibility of observing electron paramagnetic resonance transitions between the ground-state doublet states, which can be suppressed in perfectly axial single-ion magnets, renders the studied systems interesting as representations of quantum bits.
Introduction
The strong magnetic anisotropy of 4f ions is an essential basis for the properties of technologically applied magnetic materials. Magnetic anisotropy is determined by the local ligand (or crystal) field and a detailed understanding is of paramount importance to achieve control over it. Within the field of molecular magnetism, single 4f ions shielded from adjacent magnetic centres, typically by organic ligands, have gained interest due to the observation of intrinsic slow relaxation of the magnetisation with energy barriers for magnetisation reversal of as much as 652 cm−1.1 Such molecular systems are commonly referred to as mononuclear single-molecule magnets (SMMs) or single-ion magnets (SIMs). Recently, promising results for the incorporation of 4f SIMs in spintronics devices2 have opened up questions regarding the sensitivity of the magnetic properties towards small perturbations unavoidable in the anchoring or deposition of the molecules to e.g. a nanotube or a substrate. The strong response of the magnetic properties to the ligand field (LF) in traditional 4f magnets also holds true for lower-dimensional, molecular magnets like 4f SIMs.3 This has initiated several approaches to estimate LF splittings by calculations from e.g. ab initio4 or charge distributions.5Ab initio calculations on 3d clusters have shown the crucial role played by the second coordination sphere.6a Using single-crystal measurements, corroborated by ab initio calculations, Sessoli and co-workers demonstrated for 4f complexes exceedingly high sensitivity of the magnetic anisotropy towards the LF and that simple magneto–structural correlations used for transition element SMMs may fail completely for predicting e.g. the direction of the easy-axis of magnetisation in 4f SIMs.1b,6 These results present a challenge for the design and control of nanostructured devices relying on 4f SIMs and point to the need for a detailed understanding of the sensitivity of the LF towards small changes of the first and second coordination sphere. Mimicking the environment experienced by a 4f SIM outside the native crystallographic environment is a complicated task. Because of the difficulties in acquiring structural information of metal ion complexes in solution or for surface-attached systems, such studies are best performed by structural modifications in the solid state. Nevertheless, even in the solid state the most detailed understanding of the magnetic properties of 4f systems is in most cases hampered by the lack of fundamental knowledge about the eigenvalues and eigenvectors of the single-centre ligand field operators. To obtain this information, the luminescence pertinent to most 4f ions has gained increasing popularity as a spectroscopic tool to estimate LF splittings in SIMs, but the successful parameter determination is significantly limited by the spectroscopic resolution, possible occurrence of “hot” transitions and non-radiative decay.1b,7,8 Furthermore, the 4f–4f luminescence is not always accessible and can be screened by strong ligand-centred optical transitions as in the case of phthalocyaninate systems. In solid-state physics, information on the LF levels in 4f systems has traditionally been acquired by inelastic neutron scattering (INS) but, surprisingly, this technique has not been used to study any LF spectra of 4f SIMs.9Here, a small class of structurally similar ErIII SIMs has been investigated by INS spectroscopy and magnetometry. The compounds differ by peripheral ligand modifications and the presence or lack of crystallographic trigonal symmetry of the 4f centre. The parent complex; Er(trensal) (1) (H3trensal = 2,2′,2′′-tris(salicylideneimino)triethylamine), which is part of an isostructural series,10 was studied by Riley and co-workers who parameterised the ligand field based on optical spectroscopy, but never reported on the magnetic properties.11
The trensal3− back-bone is relatively rigid when coordinating to lanthanide ions and can easily be functionalised.12 An attractive feature of 1 is the presence of a crystallographic three-fold symmetry of the ErIII ion (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c1 space group) and the concomitant reduced number of symmetry-allowed LF parameters. Further, high-resolution optical spectra are available from which all possible LF parameters were unraveled.11a Additionally, using the slightly modified ligand system 2,2′,2′′-tris(3-iodo-5-methylsalicylideneimino)triethylamine (3-I,5-Me-trensalH3) gives Er(3-I,5-Me-trensal) (2, cf.Scheme 1) again being trigonal (P space group). On the contrary, chlorine-substituted 2,2′,2′′-tris(5-chlorosalicylideneimino)triethylamine) (5-Cl-trensalH3) yields Er(5-Cl-trensal) (3), which crystallises in the monoclinic P21/c space group with no axial, local symmetry. For these reasons, the presented compounds are ideal test beds for the systematic study of LF perturbations induced by minute modifications of the LF geometry and strength.
c1 space group) and the concomitant reduced number of symmetry-allowed LF parameters. Further, high-resolution optical spectra are available from which all possible LF parameters were unraveled.11a Additionally, using the slightly modified ligand system 2,2′,2′′-tris(3-iodo-5-methylsalicylideneimino)triethylamine (3-I,5-Me-trensalH3) gives Er(3-I,5-Me-trensal) (2, cf.Scheme 1) again being trigonal (P space group). On the contrary, chlorine-substituted 2,2′,2′′-tris(5-chlorosalicylideneimino)triethylamine) (5-Cl-trensalH3) yields Er(5-Cl-trensal) (3), which crystallises in the monoclinic P21/c space group with no axial, local symmetry. For these reasons, the presented compounds are ideal test beds for the systematic study of LF perturbations induced by minute modifications of the LF geometry and strength.
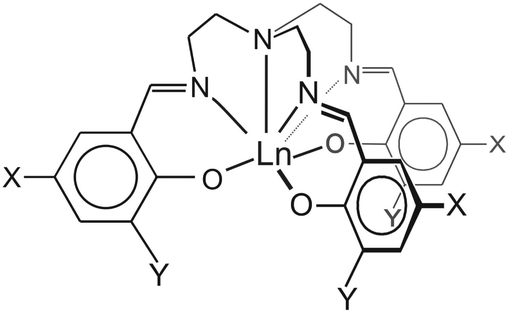 | ||
| Scheme 1 Pictorial representation of 1–3: 1 X = Y = H; 2: X = CH3, Y = I; 3: X = Cl, Y = H. The three-fold rotation axis in 1 and 2 lies along the axial, tertiary amine N–Ln bond. | ||
Experimental section
Synthesis
All starting materials were purchased from commercial sources and used without further purification. The large-scale synthesis of 1 was first performed by the procedure published by Bernhardt et al.10a However, this procedure often gave powder samples containing significant amounts of an unidentifiable phase. For this reason, we employed another, modified literature procedure of Kanesato and Yokoyama which also proved useful to obtain large single crystals suitable for single-crystal SQUID magnetometry.13 In addition, all employed samples were rigorously characterised by single-crystal or powder X-ray diffraction and elemental analysis. Er(CF3SO3)3·9H2O (0.50 g, 0.64 mmol) and tris(2-aminoethyl)amine (“tren”, 0.19 g, 1.3 mmol) were refluxed in acetonitrile (15 ml) for 15 min. The solution was cooled and placed at the bottom of a 35 ml glass tube (∅ ≈ 8 mm) and layered with acetonitrile (ca. 20 ml) and salicylaldehyde (0.24 g, 2.1 mmol). Large pencil-shaped crystals developed over a week. For 2, 3-iodo-5-methylsalicylaldehyde was synthesized as described in literature.14 For 3-I,5-Me-trensal, to 3-iodo-5-methylsalicylaldehyde (7.5 g, 29 mmol) dissolved in boiling methanol (100 ml) was added tris(2-aminoethyl)amine (1.5 g, 10 mmol). After cooling to room temperature, the crystalline, yellow product was isolated by filtration and washed with methanol. Yield: 7.8 g (88%). Calc. (found) for C30H33I3N4O3 (%): C, 41.02 (41.04); H, 3.79 (3.48); N, 6.38 (6.33). Subsequently, Er(NO3)3·5H2O (220 mg, 0.50 mmol) was dissolved in methanol (20 ml) and added to a boiling methanol (150 ml) solution of 3-I-5-Me-trensal (0.40 g, 0.46 mmol) and triethylamine (0.14 g, 1.4 mmol). The boiling was continued for 5 min, the mixture was cooled to RT and 2 was isolated by filtration and washed with methanol. Yield: 0.44 g (88%). Polycrystalline samples of 3 were synthesised as reported for the Gd analogue and proven to be isostructural from X-ray powder diffraction. The structural data for the Gd analogue with Gd replaced by Er was employed in the ab initio calculations for 3.15 Recrystallisation from boiling methanol afforded single crystals suitable for structure determination (cf. Table S1 and Fig. S3†). This phase is found to be different from the powdered sample used in the INS and magnetic measurements and no further studies were performed on this phase. The diamagnetic Y analogues (1′–3′) were synthesized similarly and shown by X-ray powder diffraction to be isostructural to the Er systems (cf. Fig. S4–6†). Er-doped 1′ samples were obtained similarly. Elemental analysis results (%): Calc. (found) for 1: C, 52.07 (52.01); H, 4.37 (4.01); N, 9.00 (8.98). Calc. (found) for 2: C, 34.56 (34.86); H, 2.90 (2.60); N, 5.37 (5.38). Calc. (found) for 3 (C27H24Cl3ErN4O3): C, 44.66 (44.53); H, 3.33 (2.95); N, 7.72 (7.65). Calc. (found) for 1′ (C27H27N4O3Y): C, 59.56 (58.86); H, 5.00 (4.95); N, 10.29 (10.52). Calc. (found) for 2′: C, 37.37 (37.47); H, 3.14 (2.79); N, 5.81 (5.77). Calc. (found) for 3′: C, 50.06 (49.90); H, 3.73 (3.40); N, 8.64 (8.58).X-Ray crystallography
Single-crystal X-ray diffraction studies were performed at 122(1) K on a Nonius Kappa CCD area-detector diffractometer (equipped with an Oxford Cryostreams low-temperature device, using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) or on a Bruker D8 VENTURE diffractometer equipped with Mo-Kα high-brilliance IμS (micro-source) radiation (λ = 0.71073 Å), a multilayer X-ray mirror and a PHOTON 100 CMOS detector, and an Oxford Cryosystems low-temperature device. The instrument was controlled with the APEX2 software package. The structures were solved using direct methods (SHELXS97) and refined using the OLEX2 programme.16 All non-hydrogen atoms were refined anisotropically, whereas H-atoms were isotropic and constrained. Crystal structure and refinement data for 1 (122 K), 2 (122 K) and 3 (122 K, recrystallised phase) are summarised in Table S1.† Powder X-ray crystallographic data were collected on a Ge Bruker D8 Advance Powder diffractometer operating in 2θ–θ configuration using Co-Kα (λ = 1.7902 Å) radiation.Elemental analysis
Elemental analysis for C, H and N was performed with a CE Instrument: FLASH 1112 series EA, at the microanalysis laboratory, University of Copenhagen.Magnetic measurements
Inelastic neutron scattering
INS spectra were obtained on the time-of-flight spectrometers IN4 and IN5 located at Institut Laue-Langevin, Grenoble, France. Several grams of non-deuterated samples were measured in double-wall Al/Mg sample cans or as wrapped in Al foil at selected temperatures and incoming neutron wavelengths. The YIII analogues 1′, 2′ and 3′ were systematically measured with the same settings to facilitate a precise assignment of the phonon spectra. The data were reduced and analysed using the Large Array Manipulation Program (LAMP).17 Magnetic (de)excitations were localised by their characteristic |Q| (linear momentum transfer) dependence which follows the magnetic form factor, their temperature variation and the comparison with the spectra of the diamagnetic YIII analogues.Electron paramagnetic resonance
EPR spectra were acquired on a Bruker Elexsys E500 equipped with a Bruker ER 4116 DM dual mode cavity, an EIP 538B frequency counter and a ER035M NMR Gauss-meter. The spectra were simulated or fitted using home-written software.18 Large single crystals of ErIII doped into 1′ were obtained as described for 1 but employing Y(CF3SO3)3·9H2O with a 5 mol% presence of Er(CF3SO3)3·9H2O.Modelling
In order to compare the LF obtained from the published optical spectra11a as well as that from ab initio calculations we will use sets of extended Stevens operator coefficients or, in short, Stevens parameters. This involves a reduction of the quantum-mechanical basis set to the states of the ErIII ground-state multiplet 4I15/2. Such a procedure is justified because the temperature range used in our measurements lies far below the energies of the first excited 4I13/2 multiplet (∼6000 cm−1).The following procedure was applied to convert the published LF parameterisation into Stevens parameters: The energy spectrum of 1 was calculated using the published LF model and best-fit parameters11a by using the same software written by M. F. Reid.19 The eigenvalues and eigenvectors in the |L,S,J,m〉 basis using 284 states at energies of up to ∼50000 cm−1 were exported into a MATLAB program.20 The LF Hamiltonian HLF in matrix form was obtained by back transformation HLF = AHdiagA† with A the unitary matrix containing the eigenvectors as columns and Hdiag the diagonal matrix of the corresponding eigenvalues. To obtain the zfs Hamiltonian only the coefficients in the eigenvectors vi referring to the ground-state multiplet 4I15/2 were considered and the resulting new eigenvectors were renormalized to norm(vi) = 1.
As mentioned later in the text least-squares fits were performed by minimising the sum of weighted and squared deviations 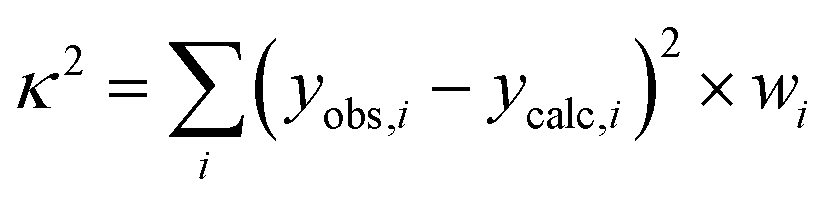 with yobs,i and ycalc,i the experimental and calculated values of the χT(T) product, field-dependent magnetisation and of the energies of the INS transitions. The weight wi results from the inverse square of the estimated experimental errors. In the case of 1 also the published energy spectrum was included as observations and a smaller weight (wi = 0.03 vs. 0.1) was given to the high-energy part of the spectrum which could not be observed by INS.
with yobs,i and ycalc,i the experimental and calculated values of the χT(T) product, field-dependent magnetisation and of the energies of the INS transitions. The weight wi results from the inverse square of the estimated experimental errors. In the case of 1 also the published energy spectrum was included as observations and a smaller weight (wi = 0.03 vs. 0.1) was given to the high-energy part of the spectrum which could not be observed by INS.
![[small tau, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b8.gif) zHz + g⊥xHx + g⊥yHy) the zfs is omitted and the magnetic anisotropy is now present in the g-factor. has the properties of a spin-1/2 angular momentum mapped on to the lowest Kramers doublet. The coordinate frame is chosen to be identical with that of the zfs Hamiltonian eqn (1) (vide infra), i.e., the z-axis coincides with the molecular C3 axis.
zHz + g⊥xHx + g⊥yHy) the zfs is omitted and the magnetic anisotropy is now present in the g-factor. has the properties of a spin-1/2 angular momentum mapped on to the lowest Kramers doublet. The coordinate frame is chosen to be identical with that of the zfs Hamiltonian eqn (1) (vide infra), i.e., the z-axis coincides with the molecular C3 axis.
All spin-Hamiltonian calculations shown in this work are based on full diagonalisation. The matrices of the extended Stevens operators were generated with the help of the stev function from the EasySpin package written by Stefan Stoll.21 The powder average of the magnetisation for large fields was generated using a 110-point Lebedev–Laikov grid.22
Results and discussion
The condensation products of tris(2-aminoethyl)amine (“tren”) with aromatic aromatic aldehydes have been extensively employed as ligands for single lanthanide ions12 and bicompartmental systems.27 However, only few of the systems possess the high symmetry that is beneficial for the present type of study. For the unsubstituted, pristine 1, the crystal structure at room temperature was reported by Kanesato and Yokoyama and our redetermination of the structure at 122 K revealed only small differences with respect to the 300 K structure. The structure is shown in Scheme 1 and Fig. 1 (Fig. S1†). 1 crystallises in the Pc1 space group and possesses crystallographic three-fold (C3 point group) symmetry of the seven-coordinate ErIII ion. For the novel 2 and 3, the space groups are P and P21/c, respectively, and the Er sites have local C3 and C1 symmetries. The bond lengths and angles of the first coordination sphere are almost identical for 1–3 as visible in Fig. 1 (Fig. S1–3†). The root-mean-square deviations of the central ErIII ion and the first coordination sphere are 0.082 Å (1vs.2) and 0.17 Å (1vs.3), respectively, illustrating the strong similarity of the first coordination spheres. Importantly, the Er–Namine distances of 2.67–2.70 Å the average Er–Nimine of 2.46–2.47 Å, and the Er–Ophenolate of 2.18–2.19 Å are all in the typical range of Ln–N and Ln–O distances. Hence the results of the present study can be considered representative for these ligators.
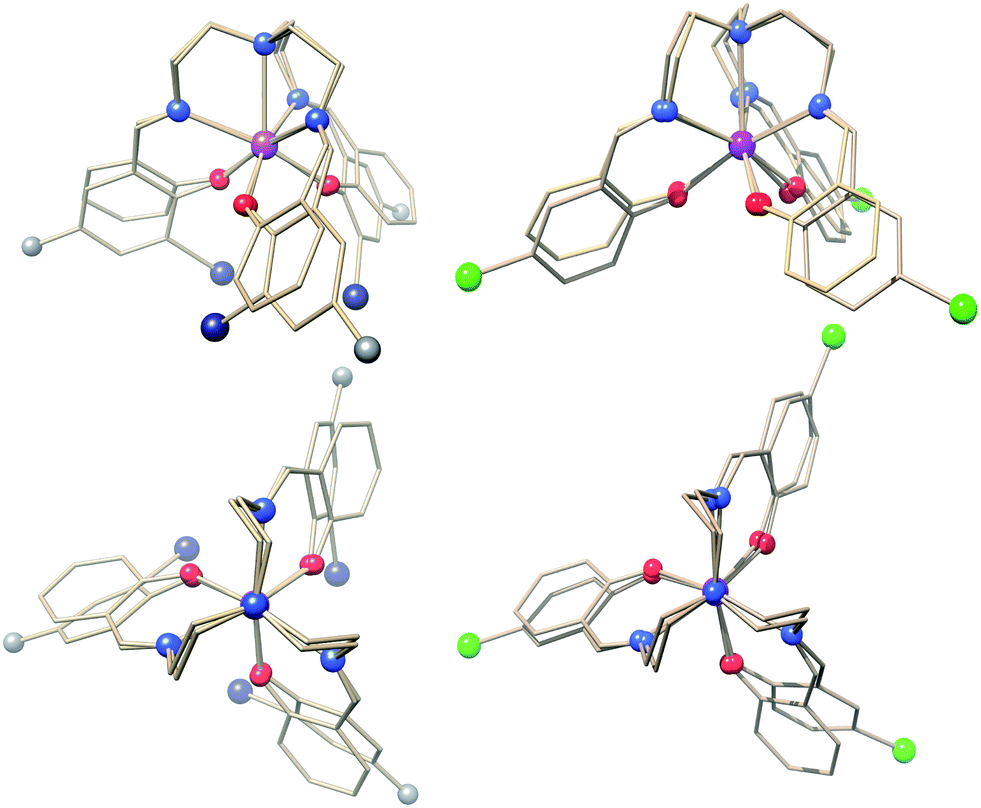 | ||
| Fig. 1 Structural overlays of 1 with 2 (left figures) and 3 (powder phase, right figures). The projections at the bottom are along the three-fold rotation axis of 1. Colour code: Er, purple; I, dark blue; Cl, green; O, red; N, blue; C, grey. With the exception of the substituents, the remaining parts of the trensal3− ligands are shown as wireframe for clarity. | ||
The ground state of ErIII is well described by a 4I15/2 Russell–Saunders term. In the absence of any symmetry the enormous number of 27 LF parameters is allowed to be nonzero and the precise determination of a unique parameter set is certainly impossible. In the presence of three-fold rotational symmetry (C3), the number of symmetry-allowed terms drops dramatically to 9. However, magnetisation and susceptibility data on most 4f systems, especially on polycrystalline samples, are not very distinct, hence the unambiguous determination of 9 parameter values still remains a challenge. For compound 1, high-resolution optical spectra are available from which all possible LF parameters were unraveled by Riley and co-workers.11a The LF parameters were fitted to several multiplets, however, here we are concerned with the magnetic properties which are largely determined by only the ground multiplet. Hence all excited multiplets can be safely neglected. This can be understood by considering that the first excited 4I13/2 multiplet is separated by more than 6000 cm−1 from the ground state while the full span of the LF-split 4I15/2 ground multiplet is around 600 cm−1.11a The limited resolution of the optical spectroscopy and approximations that result from the translation of LF into zero-field splitting (zfs) parameters lead to only a rough prediction of the magnetic properties which are extremely sensitive to small changes in the LF. Therefore, an optimised set of zfs parameters for the ground-state multiplet of 1 is sought by fitting the zfs Hamiltonian simultaneously to the neutron and optical spectroscopic data as well as to the temperature and field dependence of the magnetisation.
In order to compare the ab initio calculated LF with that from optical spectroscopy both were translated into an extended Stevens operator description working on the 4I15/2 ground multiplet
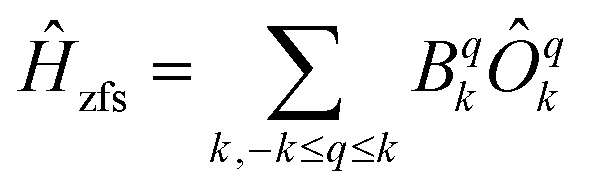 | (1) |
Magnetic field was taken into account in the phenomenological LF model by the effective Zeeman Hamiltonian Ĥz = gErμ0μBĴ·H while an exact microscopic expression has been used in ab initio calculations.4 From Ĥ = Ĥz + Ĥzfs the magnetic properties and the INS spectra were calculated. Further, least-squares fits to the magnetic data and the neutron spectra were performed to find optimised parameter sets. In the fits, the B−34 parameters were fixed to zero which corresponds to a fixing of the coordinate frame hence it can be done without loss of generality. To allow for a comparison, the ab initio calculated parameter sets and that obtained from optical spectroscopy were transformed by rotations of the coordinate frame in order to give B−34 = 0.
Static properties of 1
The INS excitation spectra for 1 and 1′ obtained at 1.5 K are shown in Fig. 2 and S7–12.† For 1, the spectrum is dominated by two prominent transitions labelled as P1 and M1 and located at 31 and 53 cm−1, respectively. The peaks are intense and nicely visible although the experiments were performed on non-deuterated samples. The phonon background can be estimated by downscaling a high-temperature spectrum according to the Bose factor28 [1 − exp(−ℏω/kBT)]−1 as shown in Fig. 2. Phononic and magnetic peaks are distinguished by studying the dependence of their intensity on temperature (Fig. S9–11†), on linear momentum transfer Q (Fig. S7 and 8†), and by comparison with the estimated phonon background and with the purely phononic spectrum of the nonmagnetic 1′. Accordingly, phononic and magnetic features are labelled as Pn and Mn, respectively. The temperature dependence of M1-3 indicates that the associated transitions are excitations from the ground state to excited states (“cold transitions”). Depending on the nature of the phonon modes, their energy is a function of the mass of the involved 4f ion, thereby explaining the small difference between the spectra of 1 and 1′ around the feature P1. The positions of M1, M2 and M3 of 53, 102 and 111 cm−1 are in perfect agreement with the energies reported by Flanagan et al. of 54, 102 and 110 cm−1.11a Given the precise knowledge of the energy spectrum it is interesting to perform a correlation with the magnetic properties. Also this provides one of the rare opportunities to further examine the performance of the LF parameters reported by Flanagan et al. as well as state-of-the-art CASSCF/RASSI-SO calculations. The ab initio calculated crystal field components of the J = 15/2 multiplet in 1 taking into account the structure obtained at T = 293 K are given in Table 1. The last column indicates the multiplet energies extracted from luminescence spectra. The agreement with ab initio calculated energies is remarkably good for the lowest four Kramers doublets (cf.Fig. 2). Also the calculated energies arising from higher multiplets (J = 13/2, 11/2) compare very well (cf.Table 1). Further we have employed the experimental structure determined at low temperature (T = 122 K) for similar ab initio calculations. The resulting energy spectrum (Table S3†) and extended Stevens parameters (Table S4†) are similar to those obtained for the room-temperature structure.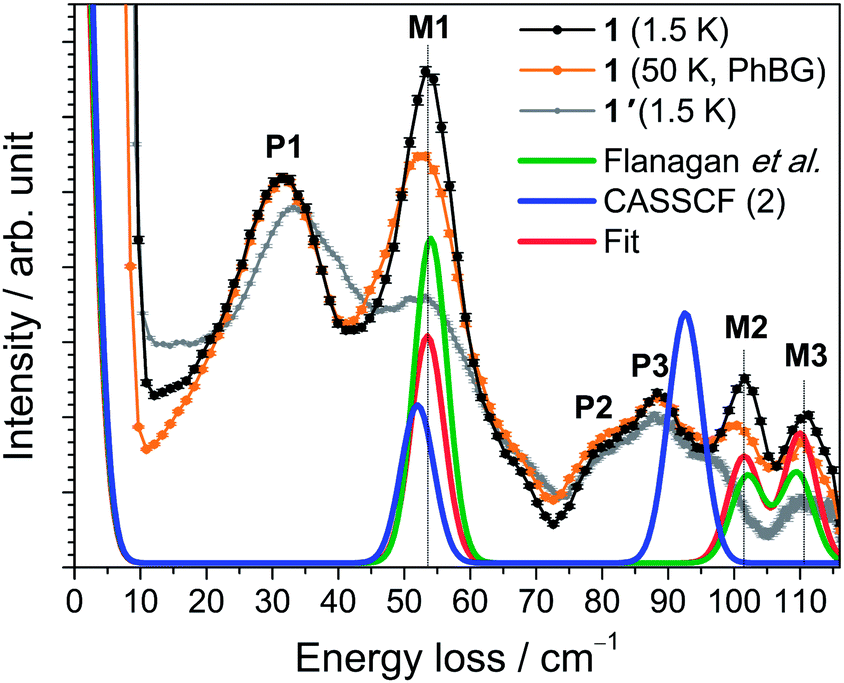 | ||
| Fig. 2 INS spectra of 1 (black trace) and 1′ (grey) acquired with λi = 2.2 Å at T = 1.5 K. The intensity was summed over the complete available Q range. The calculated phonon background from the 50 K spectrum (see main text) is shown as an orange curve (PhBG). The lower red, blue and green traces are simulations employing different sets of Stevens parameters as described in the text. | ||
| Free-ion multiplet | One molecule CASSCF (1) | One molecule embedded in 5 layers of point charges CASSCF (2) | Observed (ref. 11a) |
|---|---|---|---|
| 4I15/2 | 0 | 0 | 0 |
| 64 | 52 | 54 | |
| 99 | 91 | 102 | |
| 103 | 93 | 110 | |
| 198 | 203 | 299 | |
| 421 | 410 | 568 | |
| 459 | 448 | 610 | |
| 484 | 472 | 642 | |
| 4I13/2 | 6652 | 6650 | 6594 |
| 6687 | 6679 | 6612 | |
| 6692 | 6683 | 6621 | |
| 6705 | 6705 | 6690 | |
| 6903 | 6892 | 6909 | |
| 6904 | 6893 | 6928 | |
| 6917 | 6906 | 6939 | |
| 4I11/2 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 716 716 |
10711 |
10291 |
| 10724 |
10722 |
10301 |
|
| 10738 |
10732 |
10316 |
|
| 10863 |
10854 |
10444 |
|
| 10869 |
10859 |
10449 |
|
| 10883 |
10872 |
10510 |
|
The magnetic properties of 1 measured as field-dependent magnetisation M(H) and product of magnetic susceptibility and temperature (χT) on a polycrystalline sample and on a single crystal parallel and perpendicular to the crystallographic three-fold axis are shown in Fig. 3 and 4. The temperature dependence of the magnetisation and the corresponding fits using Hamiltonian eqn (1) are shown in Fig. S25.†
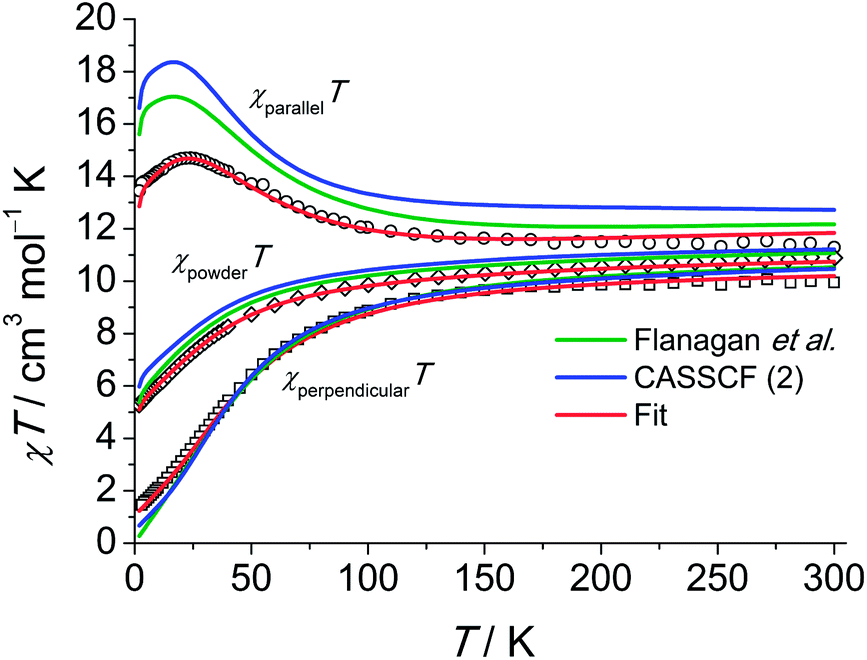 | ||
| Fig. 3 Experimental χT (χ = M/H, H = 2000 Oe) products for a single crystal of 1 along (○) and perpendicular (□) to the C3 axis and for a polycrystalline sample of 1 (◇, H = 1000 Oe). For the polycrystalline sample the χT was calculated as (χxxT + χyyT + χzzT)/3. Calculations using eqn (1) are shown as solid lines as indicated in the plot. | ||
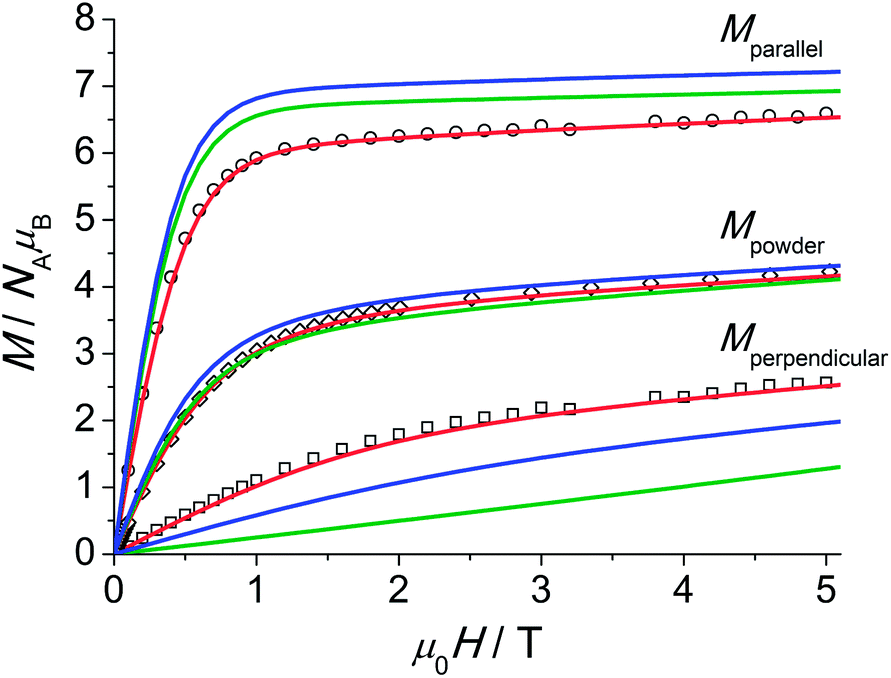 | ||
| Fig. 4 Experimental magnetisation at T = 2.0 K for 1 along (○) and perpendicular (□) to the C3 axis and for a polycrystalline sample (◇). The powder average of the magnetisation (M) was obtained by averaging over a spherical surface. The colour labelling of the simulations and fit is identical to that employed in Fig. 3. | ||
The single-crystal magnetisation data are clearly proving the presence of strong magnetic anisotropy. Further, at 300 K the χT exhibits still a distinct anisotropy confirming the overall span of the LF split ground multiplet being comparable to the thermal energy at room temperature. The X-band EPR spectra of an ErIII-doped 1′ (ca. 5 mol%) single-crystal provide g|| = 11.8 and g⊥ = 3.53 (cf. Fig. S28†) in excellent agreement with the low-temperature part of the χT data. Furthermore, the observation of an EPR signal from the ground-state doublet directly confirms that the ground-state Kramers doublet transforms like the Γ4,5 irreducible representation29 as stated in ref. 11a. The calculation of the magnetisations parallel and perpendicular to the three-fold axis using the effective spin-1/2 Hamiltonian with the g-values from EPR slightly underestimates the magnetisation compared to the experimental data (cf. Fig. S26†). This is particularly severe at larger field due the admixture with excited states leading to a change in the wavefunctions (second-order effects). The fitted Stevens parameter set for 1 using the zfs Hamiltonian eqn (1) yields nearly perfect agreement with all magnetic and spectroscopic data. The effective g-factors of the lowest Kramers doublet derived from the fitted Stevens parameters are g|| = 11.9 and g⊥ = 3.36 which coincide almost perfectly with the values found from EPR. The ab initio calculated main values are g|| = 13.68 and g⊥ = 2.28. The g-factor anisotropy is somewhat overestimated, which can be due to the following reason: the first excited Kramers doublet possesses opposite magnetic anisotropy with respect to the ground one, being of easy plane type (g|| = 2.87 and g⊥ = 7.62). This means that small modifications of the LF will strongly influence the magnetic anisotropy of the ground multiplet given the proximity of the first excited Kramers doublet. The calculations employing the low-temperature structure show a similar trend (Table S5†).
An overview of the Stevens parameters obtained for 1 by ab initio calculations, from optical spectroscopy and from fits to the magnetic and INS data is given in Table 2. There is good agreement between the different parameter sets. Parameters forbidden in C3 symmetry come out to be non-zero from ab initio calculations because of limited numerical accuracy due to employment of Cholesky decomposition of the bielectronic integrals,23 however, they are small compared to most of the allowed parameters of the same rank k. Since the comparison of sets of Stevens parameters is rather abstract it is of great help to examine the performance of the ab initio calculations and LF parameters from optical spectroscopy in reproducing the magnetic and INS data. Notably, as visible from the magnetisation and susceptibility data, the parameter sets obtained from optical spectra and ab initio calculations suggest slightly stronger anisotropy than what is consistently obtained from single-crystal magnetic data and EPR measurements.
| Compound 1 | Compound 2 | Compound 3 | ||||
|---|---|---|---|---|---|---|
| k, q | ab initio CASSCF (2) | Flanagan et al.11a | Best fit | ab initio CASSCF (2) | Best fit | Best fit |
| 2, −2 | 3.780 × 10−3 | 3.244 × 10−3 | ||||
| 2, −1 | −5.321 × 10−3 | 6.266 × 10−3 | ||||
| 2, 0 | −0.8776 | −0.975 | −1.07(2) | −2.824 × 10−1 | −1.9(6) × 10−1 | 1.22(2) |
| 2, 1 | 5.058 × 10−3 | −3.124 × 10−3 | ||||
| 2, 2 | 3.439 × 10−4 | 1.775 × 10−3 | ||||
| 4, −4 | 1.274 × 10−4 | −1.964 × 10−4 | ||||
| 4, −3 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4, −2 | 1.273 × 10−4 | −1.894 × 10−4 | ||||
| 4, −1 | −7.482 × 10−5 | 1.359 × 10−4 | ||||
| 4, 0 | −1.010 × 10−3 | −0.260 × 10−3 | −0.2(2) × 10−3 | −1.461 × 10−3 | −6.0(6) × 10−3 | −3.9(5) × 10−3 |
| 4, 1 | 6.359 × 10−5 | −2.101 × 10−5 | ||||
| 4, 2 | 8.629 × 10−5 | −3.777 × 10−5 | ||||
| 4, 3 | 0.1770 | 0.287 | 0.255(5) | 1.707 × 10−1 | 1.1(4) × 10−1 | 1(1) × 10−2 |
| 4, 4 | −1.121 × 10−4 | 1.348 × 10−5 | ||||
| 6, −6 | 1.555 × 10−5 | 5.89 × 10−4 | 3.5(5) × 10−4 | 4.235 × 10−5 | 1.6(7) × 10−4 | 1.2(1) × 10−4 |
| 6, −5 | 1.335 × 10−5 | −4.616 × 10−6 | ||||
| 6, −4 | −4.404 × 10−7 | 5.479 × 10−7 | ||||
| 6, −3 | −1.165 × 10−4 | −2.45 × 10−4 | −1(2) × 10−4 | −2.341 × 10−4 | 1.3(2) × 10−3 | 7(7) × 10−5 |
| 6, −2 | −2.360 × 10−6 | 1.853 × 10−6 | ||||
| 6, −1 | 3.101 × 10−6 | −6.209 × 10−6 | ||||
| 6, 0 | 9.685 × 10−5 | 1.23 × 10−4 | 1.25(1) × 10−4 | 8.896 × 10−5 | 9.5(6) × 10−5 | −1.07(3) × 10−4 |
| 6, 1 | −2.971 × 10−6 | 1.485 × 10−6 | ||||
| 6, 2 | −1.892 × 10−6 | −1.123 × 10−6 | ||||
| 6, 3 | −0.8741 × 10−3 | −0.943 × 10−3 | −1.27(2) × 10−3 | −5.176 × 10−4 | −1(5) × 10−5 | 3.13(8) × 10−6 |
| 6, 4 | 8.189 × 10−7 | −8.547 × 10−7 | ||||
| 6, 5 | 7.960 × 10−6 | 4.747 × 10−6 | ||||
| 6, 6 | 0.8663 × 10−3 | 1.03 × 10−3 | 1.27(2) × 10−3 | 7.316 × 10−4 | 4.2(8) × 10−4 | 1.4(1) × 10−5 |
| g Er | 6/5 | 6/5 | 1.18(1) | 6/5 | 1.18(1) | 1.18(1) |
Static properties of 2
The INS spectra of 2 and the dc magnetic data are shown in Fig. 5 and 6, respectively. The detailed temperature dependence of the INS spectra of 2 and 2′ is given in Figs. S13–17.† The neutron spectra of 2 (Fig. 5) are remarkably different from those of 1. The magnetic and phononic features were disentangled using the same criteria and procedures as for 1. In the 2.8 Å spectra, a prominent peak, M2, is observed at 70 cm−1 and a weaker feature M1 at 37 cm−1. In the 1.4 Å spectrum the magnetic features are not very strong. An additional magnetic feature M3 at an energy loss of 140 cm−1 can be identified (cf. Fig. S13†).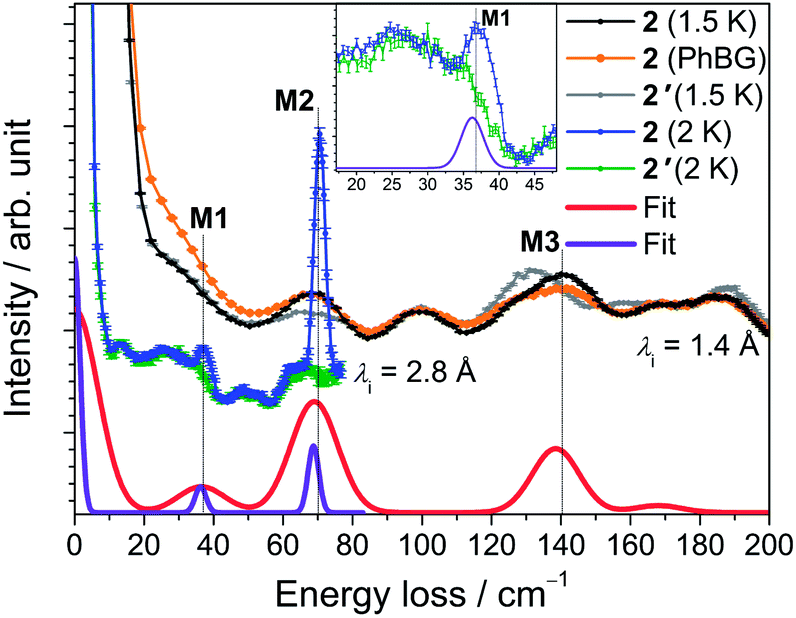 | ||
| Fig. 5 INS spectra of 2 (black and blue traces) and 2′ (grey and green traces) obtained with incident neutron wavelengths of 1.4 Å (at T = 1.5 K) and 2.8 Å (at T = 2 K). The curves are offset to improve clarity. The calculated phonon background from the 40 K spectrum is shown as a solid orange line. The red and purple lines are simulations with resolutions comparable to the 1.4 and 2.8 Å data set, respectively, as described in the text. Inset: Zoom of the low-energy region of the 2.8 Å spectra around peak M1. | ||
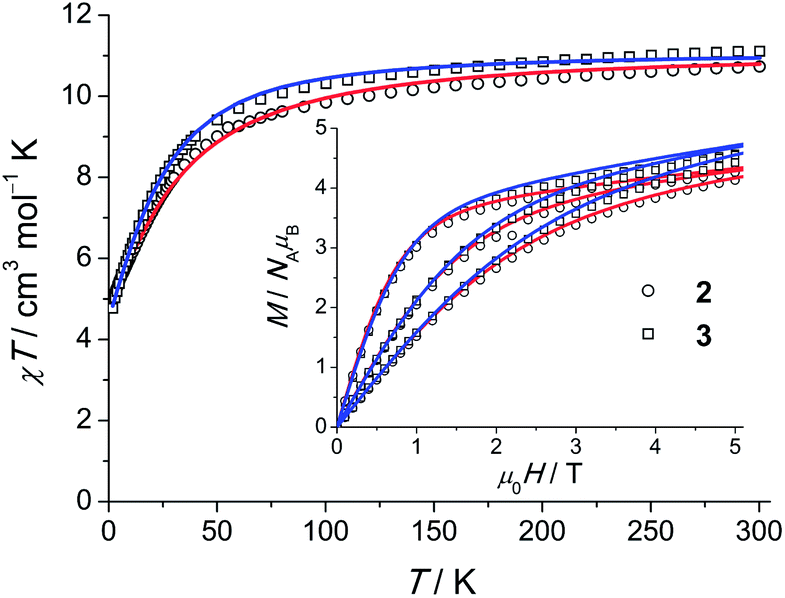 | ||
| Fig. 6 χT (χ = M/H, H = 1000 Oe) products for polycrystalline 2 and 3. Open symbols correspond to the experimental data, solid lines are calculated curves obtained from the model of eqn (1). Field-dependent magnetisation M(H) at 2, 4 and 6 K is shown in the inset. | ||
Least-squares fits of all C3-allowed Stevens parameters to the magnetic data and the INS spectra of 2 were performed as described for compound 1. The best–fit parameters and the corresponding simulated curves are shown in Table 2 and in Fig. 5 and 6. By using random initial values we were able to obtain three parameter sets only slightly differing in κ2 (cf. Modelling Section) that reproduce all available data very well. A comparison of the three sets yields that the ‘axial’ parameters B02, B04 and B06 are quite robust, while the ‘non-axial’ parameters are fluctuating. The ab initio calculated magnetic properties and INS spectra, which were based on the 122 K structural data, are presented in Figs. S32–34.† While the calculated magnetic properties are only slightly deviating from the experimental data, the INS spectra do not match well. Given that the ab initio predictions for the similar compounds 1 and 2 are expected to be of equal accuracy, the likely explanation for the larger discrepancy in 2 is that the employed 122 K structural data differs significantly from the geometry at 1.5 to 40 K, where the INS experiments were performed. In such a situation INS and optical spectroscopic data are indispensable for an accurate description of the electronic spectra of lanthanide SIMs.
Static properties of 3
For 3, a representative INS spectrum at 2.2 Å is shown in Fig. 7. More INS data of 3 and 3′ are shown in Fig. S18–22.† The comparison of the spectra of 3 and 3′ reveals magnetic excitations at 29 cm−1 (M1), 76 cm−1 (M2) and 106 cm−1 (M3). An inspection of the temperature dependence indicates that M1-3 are cold transitions. Comparing the 1.1 Å spectra indicate no further magnetic excitations up to energy transfers of ca. 500 cm−1. However, in the temperature dependence of the 2.8 Å spectra, a magnetic excitation (m4) starting out from an excited state (“hot” transition) was located at 48 cm−1. The parameters derived from ab initio calculations, done for the room-temperature structure of Gd(5-Cl-trensal)15 in which Gd has been replaced by Er, do not satisfactorily reproduce the experimental data (cf. Tables S9–11 and Fig. S35–37†). Structural changes due to the exchanged central ion, or changes not detectable in powder XRD and/or due to the low temperatures in the experiments are obvious reasons for this discrepancy. A fit to the magnetic data and INS spectra using the ab initio derived parameters as initial values stops in a local minimum. To extract LF parameters within our phenomenological model, fits using random initial values were performed. The best results are plotted as solid lines in Fig. 6 and 7. While there are several parameter sets which lead to an excellent reproduction of the M(H) and χT, the INS plays a decisive role here. Only one of the parameter sets reproduces well also the warm INS transition m4 (cf. Fig. S20†). In these fits, only parameters allowed in C3 symmetry were considered, which is certainly a strong approximation in view that this compound actually possesses lower symmetry. However the introduction of more fit parameters would not lead to a significant improvement of the fits since a good fit can already be achieved with the current parameter set. In such a situation, in order to find values of all 27 LF parameters ab initio calculations are indispensable. Our study shows that the ligand field spectrum of lanthanides is highly sensitive to the structural details. These may not be correctly captured by the structures of isostructural compounds as emphasized by the example of compound 3.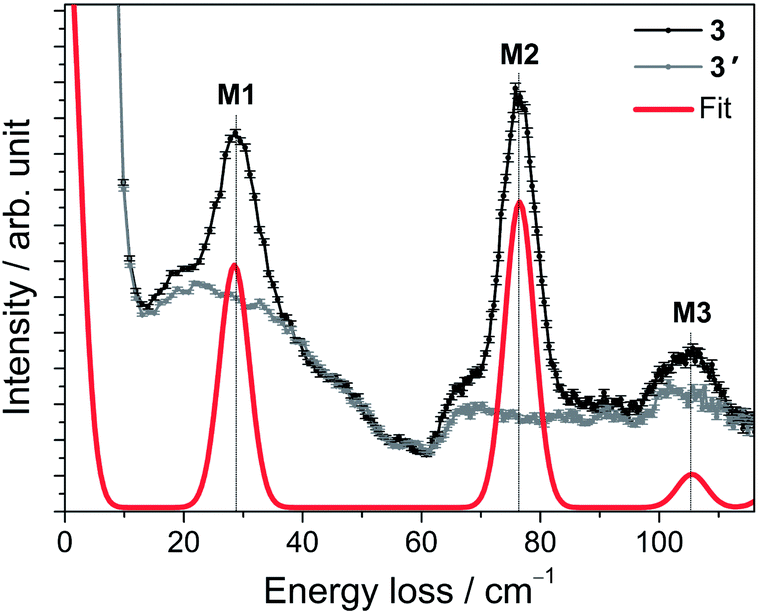 | ||
| Fig. 7 INS spectra of 3 (black trace) and 3′ (grey trace) acquired with an incident neutron wavelength of 2.2 Å at T = 1.5 K. The red line is the simulation using the best-fit parameters as described in the text. | ||
Dynamic magnetic properties of 1–3
The temperature dependencies of the magnetisation relaxation times of 1 (polycrystalline and as oriented single-crystal), Er-doped 1′ and 3 measured by ac SQUID magnetometry are shown in Fig. 8. In the absence of a static field none of the compounds 1–3 showed a peak in the out-of-phase component (χ′′) of the ac susceptibility (Fig. S42, S47 and 48†). In small dc fields, clear maxima in χ′′(νac) are observed for 1 and 3, whereas only onsets of peaks are observed in 2 with νac ≤ 1.5 kHz at 1.8 K (cf. Fig. S47†). The magnetisation curves of an aligned (magnetic field applied parallel to the C3 axis) single crystal were also measured using a micro-Hall sensor at temperatures down to 1.4 K and magnetic field sweep rates up to 2.3 T s−1. However, magnetic hysteresis was not observed (cf. Fig. S51 and 52†). A polycrystalline sample of 10% ErIII doped into 1′ shows almost the same temperature dependence as pristine 1. The field dependence (cf. Fig. S50†), however, hints at a weak effect of intermolecular interactions, which are suppressed in the diluted sample. For this reason we focus here on the latter compound, in order to investigate the mechanism that dominates the magnetisation relaxation.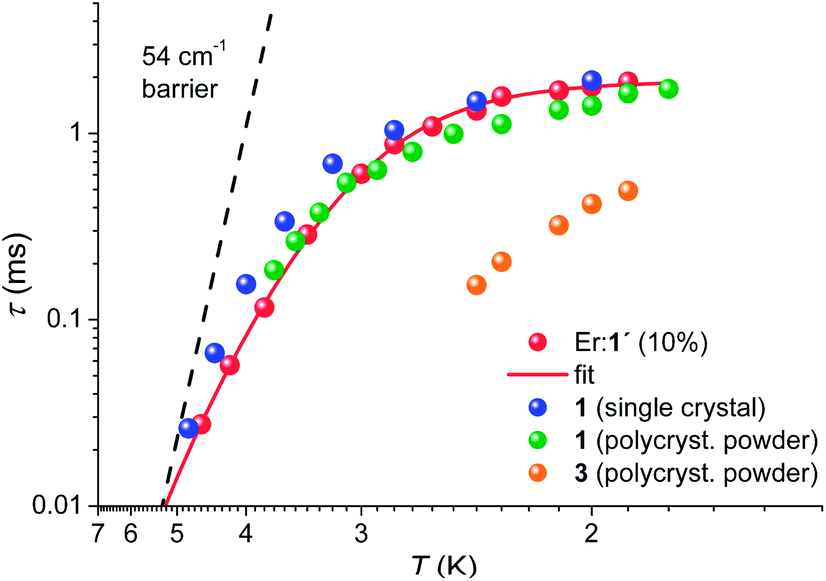 | ||
| Fig. 8 Arrhenius plot for 1 as a single crystal measured with Hdc = 900 Oe applied along the C3 axis, as polycrystalline material (Hdc = 900 Oe), and as Er doped into 1′ (Er:1′), Hdc = 1100 Oe. For 3, the data were acquired on a polycrystalline sample only (Hdc = 700 Oe). The solid red line represents the best fit to the Er:1′ data using the model of eqn (2). The dashed solid line is the Arrhenius prediction τ(T) = 9ps × exp[54 cm−1/(kBT)]. | ||
With the frequently made assumption of an Orbach process and a temperature-independent quantum tunnelling of magnetisation (QTM) as dominant relaxation processes, a good fit can be obtained yielding a barrier for magnetisation reversal of Δeff = 20(1) cm−1. The size of this barrier is, however, not compatible with the observed excited-state separation of 54 cm−1 and an Orbach process can be ruled out as a dominating relaxation mechanism in the studied temperature range of 1.9 K < T < 5 K. In view of this narrow temperature range and the gradually increasing slope towards higher temperatures it might however be possible that the Orbach process becomes the leading mechanism at elevated temperatures. It has been proposed theoretically and experimentally that first and second-order Raman processes may be significant.30 Indeed we find that the relatively simple model for the temperature-dependent magnetisation relaxation times, eqn (2):
| τ−1 = τQTM−1 + CTn | (2) |
When comparing the magnetic and spectroscopic properties of compounds 1–3 studied in this work, it becomes clear that they are vastly different. This is also reflected in the results of the ab initio calculations. Interestingly, the introduction of substituent groups far from the first coordination sphere leads to drastic modifications of the low-lying energy spectrum, hence the changes are not at all just a small perturbation to the system. The strong effect of the second coordination sphere on the anisotropy of the metal ion in a CoII2(calix-8-arene)2 complex was found by ab initio calculations to originate from the significantly stronger chemical bond within the ligand group than between the ligand atoms and the metal ion.6a Accordingly, in lanthanide complexes the sensitivity of the magnetic anisotropy to modifications in the second (and more distant) coordination sphere can be expected to be even higher. As depicted in Fig. 1 the first coordination sphere of the series 1–3 is largely similar. Therefore, the differences in the properties are likely to arise from two contributions. One comes from the difference in the electron withdrawing or donating effects imposed by the substituents and another from the subtle structural distortions of the first and second coordination sphere. The latter contributions are likely to be associated with changes in π interactions that are expected to be strongly dependent on the tilting of the aromatic rings. As an evidence for this picture, changes in the first coordination sphere have induced strong changes in the magnetic behaviour for TbIII-phthalocyaninate systems, where a contraction of the ligand sphere lead to an increase of the barrier for magnetisation reversal.31 A few recent studies further suggest that subtle changes in the far range can also have strong effects on lanthanide anisotropy.32 A strong response of the 57Fe Mössbauer spectra was detected in a Fe2Dy2 SMM upon peripheral ligand substitution suggesting a concomitant change of the lanthanide anisotropy and 3d–4f exchange coupling.33 Moreover, similar effects were proposed on the basis of an ab initio study on the SIM Na[Dy(DOTA)]·4H2O.1b These recent studies already shed some light onto this topic but it was so far difficult to imagine that the spectroscopic changes were so profound.
These results should have an impact on the current efforts of studying surface-deposited SMMs and SIMs and the grafting of such species on to carbon nanotubes and other nanostructured materials. It certainly cannot be taken for granted that the magnetic properties remain unchanged after the grafting or surface deposition process even if the molecular stoichiometry is retained and the molecules seem to be intact. Our study suggests that even if the first coordination sphere remains unchanged after the surface deposition, the interaction of the ligand with the surface may have similar effects as the ligand functionalisation studied here. However, it should also be noted that the present results can be seen as an opportunity to use the surface in order to modify the magnetic properties of the deposited SMMs or SIMs. Currently, this still seems to be out of reach mainly because the understanding of the observed effects is underdeveloped. Controlling and harnessing such interactions may open up paths to achieving unprecedented spintronic applications.
Conclusions
In summary, an extensive study of the magnetic and low-energy spectroscopic properties of a small family of ErIII SIMs has been performed. The measured data are compared to results from previously published optical spectroscopy as well as to state-of-the-art ab initio CASSCF/RASSI-SO calculations. The peripheral ligand functionalisation leads to largely unexpected drastic changes in all properties. These results are important in view of the on-going grafting of SMMs and SIMs on to surfaces and nano-objects suggesting that changes far away from the first coordination sphere can lead to vastly different magnetic properties. If control over these effects can be achieved, it may open a path to obtaining superior magnetic properties by using advantageous combinations of SIMs and surfaces.Moreover, strong evidence is delivered that the Orbach mechanism is of minor importance for the relaxation of magnetisation in these systems at temperatures 1.9 K < T < 5 K. In contrast, it appears that the relaxation is dominated by tunnelling of magnetisation at the lowest temperatures and that two-phonon Raman processes are dominating at temperatures close to 4 K.
Note added after submission
We became aware of an accepted manuscript by E. Lucaccini et al. about Er(trensal) and Dy(trensal) complexes.34Acknowledgements
We thank Dr M. Medarde-Barragan for help with the PPMS measurements. J. D. acknowledges funding by the Ambizione program of the Swiss National Science Foundation. K. S. P. thanks the Danish Ministry of Science, Innovation and Higher Education for an EliteForsk travel scholarship. J. B. thanks the Carlsberg Foundation and the Villum Foundation for support for diffraction equipment. H. W. and J. B. acknowledge support from the Danish Research Council FNU (12-125226). L. U. is a postdoc of the FWO-Vlaanderen (Flemish Science Foundation). Methusalem and INPAC projects of the KU Leuven are also gratefully acknowledged.References
- (a) N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 CrossRef CAS PubMed; (b) G. Cucinotta, M. Perfetti, J. Luzon, M. Etienne, P. E. Car, A. Caneschi, G. Calvez, K. Bernot and R. Sessoli, Angew. Chem., Int. Ed., 2012, 51, 1606 CrossRef CAS PubMed; (c) M. A. AlDamen, S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, C. Martí-Gastaldo, F. Luis and O. Montero, Inorg. Chem., 2009, 48, 3467 CrossRef CAS PubMed; (d) S.-D. Jiang, B.-W. Wang, G. Su, Z.-M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448 CrossRef CAS PubMed; (e) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS PubMed; (f) S.-D. Jiang, B.-W. Wang, H.-L. Sun, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2011, 133, 4730 CrossRef CAS PubMed; (g) S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, A. Camón, M. Evangelisti, F. Luis, M. J. Martínez-Pérez and J. Sesé, J. Am. Chem. Soc., 2012, 134, 14982 CrossRef CAS PubMed; (h) R. Westerström, J. Dreiser, C. Piamonteze, M. Muntwiler, S. Weyeneth, H. Brune, S. Rusponi, F. Nolting, A. Popov, S. Yang, L. Dunsch and T. Greber, J. Am. Chem. Soc., 2012, 134, 9840 CrossRef PubMed; (i) N. Ishikawa, M. Sugita, N. Tanaka, T. Ishikawa, S. Koshihara and Y. Kaizu, Inorg. Chem., 2004, 43, 5498 CrossRef CAS PubMed; (j) P. E. Car, M. Perfetti, M. Mannini, A. Favre, A. Caneschi and R. Sessoli, Chem. Commun., 2011, 47, 3751 RSC; (k) S. Jiang, B. Wang, G. Su, Z. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448 CrossRef CAS PubMed; (l) G. Chen, C. Gao, J. Tian, J. Tang, W. Gu, X. Liu, S. Yan, D. Liao and P. Cheng, Dalton Trans., 2011, 40, 5579 RSC; (m) A. Watanabe, A. Yamashita, M. Nakano, T. Yamamura and T. Kajiwara, Chem.–Eur. J., 2011, 17, 7428 CrossRef CAS PubMed; (n) H. L. C. Feltham, Y. Lan, F. Klöwer, L. Ungur, L. F. Chibotaru, A. K. Powell and S. Brooker, Chem.–Eur. J., 2011, 17, 4362 CrossRef CAS PubMed; (o) K. R. Meihaus, J. D. Rinehart and J. R. Long, Inorg. Chem., 2011, 50, 8484 CrossRef CAS PubMed; (p) M. Jeletic, P.-H. Lin, J. J. Le Roy, I. Korobkov, S. I. Gorelsky and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 19286 CrossRef CAS PubMed; (q) N. F. Chilton, S. K. Langley, B. Moubaraki, A. Soncini, S. R. Batten and K. S. Murray, Chem. Sci., 2013, 4, 1719 RSC; (r) C. R. Ganivet, B. Ballesteros, G. de la Torre, J. M. Clemente-Juan, E. Coronado and T. Torres, Chem.–Eur. J., 2013, 19, 1457 CrossRef CAS PubMed; (s) R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. L. McInnes, L. F. Chibotaru and R. E. P. Winpenny, Nat. Chem., 2013, 5, 673–678 CrossRef CAS PubMed.
- (a) M. Urdampilleta, S. Klyatskaya, J. P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502 CrossRef CAS PubMed; (b) R. Vincent, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Nature, 2012, 488, 357 CrossRef CAS PubMed; (c) M. Ganzhorn, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Nat. Nanotechnol., 2013, 8, 165 CrossRef CAS PubMed.
- (a) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC; (b) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078 RSC; (c) J.-L. Liu, Y.-C. Chen, Y.-Z. Zheng, W.-Q. Lin, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M.-L. Tong, Chem. Sci., 2013, 4, 3310–3316 RSC.
- L. F. Chibotaru, in Advances in Chemical Physics, John Wiley & Sons, Inc., 2013, p. 397 Search PubMed.
- (a) J. J. Baldoví, S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño and A. Palii, Inorg. Chem., 2012, 51, 12565 CrossRef PubMed; (b) N. F. Chilton, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and A. Soncini, Nat. Commun., 2013, 4, 2551 Search PubMed; (c) O. S. Reu, A. V. Palii, S. M. Ostrovsky, P. L. W. Tregenna-Piggott and S. I. Klokishner, Inorg. Chem., 2012, 51, 10955 CrossRef CAS PubMed.
- (a) S. Petit, G. Pillet, D. Luneau, L. F. Chibotaru and L. Ungur, Dalton Trans., 2007, 4582–4588 RSC; (b) J. Luzon and R. Sessoli, Dalton Trans., 2012, 41, 13556 RSC.
- (a) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC; (b) S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189 RSC.
- (a) M.-E. Boulon, G. Cucinotta, J. Luzon, C. Degl'Innocenti, M. Perfetti, K. Bernot, G. Calvez, A. Caneschi and R. Sessoli, Angew. Chem., Int. Ed., 2013, 52, 350 CrossRef CAS PubMed; (b) K. Yamashita, R. Miyazaki, Y. Kataoka, T. Nakanishi, Y. Hasegawa, M. Nakano, T. Yamamura and T. Kajiwara, Dalton Trans., 2013, 42, 1987 RSC.
- (a) A. Furrer and O. Waldmann, Rev. Mod. Phys., 2013, 85, 367 CrossRef CAS; (b) J. Dreiser, K. S. Pedersen, C. Piamonteze, S. Rusponi, Z. Salman, M. E. Ali, M. Schau-Magnussen, C. A. Thuesen, S. Piligkos, H. Weihe, H. Mutka, O. Waldmann, P. Oppeneer, J. Bendix, F. Nolting and H. Brune, Chem. Sci., 2012, 3, 1024 RSC; (c) M. Kofu, O. Yamamuro, T. Kajiwara, Y. Yoshimura, M. Nakano, K. Nakajima, S. Ohira-Kawamura, T. Kikuchi and Y. Inamura, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 064405 CrossRef.
- (a) P. V. Bernhardt, B. M. Flanagan and M. J. Riley, Aust. J. Chem., 2000, 53, 229 CrossRef CAS; (b) P. V. Bernhardt, B. M. Flanagan and M. J. Riley, Aust. J. Chem., 2001, 54, 229 CrossRef CAS.
- (a) B. M. Flanagan, P. V. Bernhardt, E. R. Krausz, S. R. Lüthi and M. J. Riley, Inorg. Chem., 2001, 40, 5401 CrossRef CAS PubMed; (b) B. M. Flanagan, P. V. Bernhardt, E. R. Krausz, S. R. Lüthi and M. J. Riley, Inorg. Chem., 2002, 41, 5024 CrossRef CAS PubMed.
- (a) W.-K. Wong, H. Liang, J. Guo, W.-Y. Wong, W.-K. Lo, K.-F. Li, K.-W. Cheah, Z. Zhou and W.-T. Wong, Eur. J. Inorg. Chem., 2004, 829 CrossRef CAS; (b) P. Dröse and J. Gottfriedsen, Z. Anorg. Allg. Chem., 2008, 634, 87 CrossRef; (c) S. Mizukami, H. Houjou, M. Kanesato and K. Hiratani, Chem.–Eur. J., 2003, 9, 1521 CrossRef CAS PubMed; (d) S. J. Archibald, A. J. Blake, M. Schroder and R. E. P. Winpenny, J. Chem. Soc., Chem. Commun., 1994, 1669 RSC; (e) P. Dröse, J. Gottfriedsen, C. G. Hrib, P. G. Jones, L. Hilfert and F. T. Edelmann, Z. Anorg. Allg. Chem., 2011, 637, 369 CrossRef; (f) M. Kanesato, S. Mizukami, H. Houjou, H. Tokuhisa, E. Koyama and Y. Nagawa, J. Alloys Compd., 2004, 374, 307 CrossRef CAS; (g) M. Kanesato, T. Yokoyama, O. Itabashi, T. M. Suzuki and M. Shiro, Bull. Chem. Soc. Jpn., 1996, 69, 1297 CrossRef CAS.
- M. Kanesato and T. Yokoyama, Chem. Lett., 1999, 137 CrossRef CAS.
- H. Voβ, K. J. Wannowius and H. Elias, J. Inorg. Nucl. Chem., 1975, 37, 79 CrossRef.
- M. Kanesato, F. N. Ngassapa and T. Yokoyama, Anal. Sci., 2001, 17, 1359 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- LAMP, the Large Array Manipulation Program; D. Richard, M. Ferrand and G. J. Kearley, J. Neutron Res., 1996, 4, 33 CrossRef.
- Q. Scheifele, T. Birk, J. Bendix, P. L. W. Tregenna-Piggott and H. Weihe, Angew. Chem., Int. Ed., 2008, 47, 148 CrossRef CAS PubMed.
- J. J. Dallara, M. F. Reid and F. S. Richardson, J. Phys. Chem., 1984, 88, 3587 CrossRef CAS.
- MATLAB R2010a, The MathWorks Inc., Natick, MA, USA, 2010 Search PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- V. I. Lebedev and D. N. Laikov, Dokl. Math., 1999, 59, 477 Search PubMed.
- F. Aquilante, L. De Vico, N. Ferre, G. Ghigo, P.-A. Malmqvist, P. Neogrady, T. B. Pedersen, M. Pitonak, M. Reiher, B. O. Roos, L. Serrano-Andres, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224 CrossRef CAS PubMed.
- B. O. Roos, P. R. Taylor and P. E. Siegbahn, Chem. Phys., 1980, 48, 157 CrossRef CAS.
- B. O. Roos and P.-A. Malmqvist, Phys. Chem. Chem. Phys., 2004, 6, 2919 RSC.
- L. F. Chibotaru and L. Ungur, J. Chem. Phys., 2012, 137, 064112 CrossRef CAS PubMed ; SINGLE_ANISO module is described in the MOLCAS manual: http://molcas.org/documentation/manual/node95.html.
- (a) R. Rodríguez-Cortiñas, F. Avecilla, C. Platas-Iglesias, D. Imbert, J.-C. G. Bünzli, A. de Blas and T. Rodríguez- Blas, Inorg. Chem., 2002, 41, 5336 CrossRef; (b) C. Platas, F. Avecilla, A. de Blas, C. F. G. C. Geraldes, T. Rodríguez-Blas, H. Adams and J. Mahía, Inorg. Chem., 1999, 38, 3190 CrossRef CAS; (c) J.-P. Costes, F. Dahan and F. Nicodème, Inorg. Chem., 2003, 42, 6556 CrossRef CAS PubMed; (d) F. Avecilla, A. de Blas, C. Platas, T. Rodriguez-Blas, R. Bastida, A. Macias, A. Rodriguez, D. E. Fenton and J. Mahia, Chem. Commun., 1999, 125 RSC; (e) C. Platas, F. Avecilla, A. de Blas, T. Rodriguez-Blas, C. F. G. C. Geraldes, E. Toth, A. E. Merbach and J.-C. G. Bünzli, J. Chem. Soc., Dalton Trans., 2000, 611 RSC; (f) Q.-Y. Chen, Q.-H. Luo and H.-k. Fun, J. Chem. Crystallogr., 2002, 32(10), 377 CrossRef CAS.
- (a) G. Shirane, S. M. Shapiro, and J. M. Tranquada, in Neutron Scattering with a Triple-Axis Spectrometer, Cambridge University Press, Cambridge, England, 2002 Search PubMed; (b) J. Dreiser, O. Waldmann, C. Dobe, G. Carver, S. T. Ochsenbein, A. Sieber, H. U. Güdel, J. van Duijn, J. Taylor and A. Podlesnyak, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 024408 CrossRef.
- If the ground state doublet belonged to the other possible (Γ6) irreducible representation, it would contain mixtures of only mJ = ±15/2, ±9/2 and ±3/2 states hence an intra-doublet transition would be forbidden by the EPR selection rule ΔmJ = ±1 independent of the exact wavefunction.
- (a) R. Orbach, Proc. R. Soc. London, Ser. A, 1961, 264, 458 CrossRef CAS; (b) R. Orbach and M. Blume, Phys. Rev. Lett., 1962, 8, 478 CrossRef; (c) R. W. Bierig, M. J. Weber and S. I. Warshaw, Phys. Rev., 1964, 134, A1504 CrossRef CAS; (d) J. M. Zadrozny, M. Atanasov, A. M. Bryan, C.-Y. Lin, B. D. Rekken, P. P. Power, F. Neese and J. R. Long, Chem. Sci., 2013, 4, 125 RSC; (e) D. E. Freedman, W. H. Harman, T. D. Harris, G. J. Long, C. J. Chang and J. R. Long, J. Am. Chem. Soc., 2010, 132, 1224 CrossRef CAS PubMed; (f) J. D. Rinehart and J. R. Long, J. Am. Chem. Soc., 2009, 131, 12558 CrossRef CAS PubMed; (g) A. Kiel and W. B. Mims, Phys. Rev., 1967, 161, 386 CrossRef CAS.
- S. Takamatsu, T. Ishikawa, S.-y. Koshihara and N. Ishikawa, Inorg. Chem., 2007, 46, 7250 CrossRef CAS PubMed.
- F. Habib, G. Brunet, V. Vieru, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 13242 CrossRef CAS PubMed.
- V. Mereacre, A. Baniodeh, C. E. Anson and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 15335 CrossRef CAS PubMed.
- E. Lucaccini, L. Sorace, M. Perfetti, J. Costes and R. Sessoli, Chem. Commun., 2014, 50, 1648 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic details, INS spectra, magnetic data, EPR spectra, computational details. CCDC 969146–969148. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc53044b |
| This journal is © The Royal Society of Chemistry 2014 |