 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot synthesis of an inorganic heterostructure: uniform occlusion of magnetite nanoparticles within calcite single crystals†
Alexander N.
Kulak
a,
Mona
Semsarilar
b,
Yi-Yeoun
Kim
a,
Johannes
Ihli
a,
Lee A.
Fielding
b,
Oscar
Cespedes
c,
Steven P.
Armes
b and
Fiona C.
Meldrum
*a
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK. E-mail: F.Meldrum@leeds.ac.uk; Fax: +44 (0)113 343 6565; Tel: +44 (0)113 343 6414
bDainton Building, Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, Yorkshire S3 7HF, UK
cSchool of Physics and Astronomy, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
First published on 8th November 2013
Abstract
A facile one-pot method is described for the formation of novel heterostructures in which inorganic nanoparticles are homogeneously distributed throughout an inorganic single crystal matrix. Our strategy uses nanoparticles functionalised with a poly(sodium 4-styrenesulphonate)-poly(methacrylic acid) [PNaStS-PMAA] diblock copolymer as a soluble crystal growth additive. This copolymer plays a number of essential roles. The PMAA anchor block is physically adsorbed onto the inorganic nanoparticles, while the PNaStS block acts as an electrosteric stabiliser and ensures that the nanoparticles retain their colloidal stability in the crystal growth solution. In addition, this strong acid block promotes binding to both the nanoparticles and the host crystal, which controls nanoparticle incorporation within the host crystal lattice. We show that this approach can be used to achieve encapsulation loadings of at least 12 wt% copolymer-coated magnetite particles within calcite single crystals. Transmission electron microscopy shows that these nanoparticles are uniformly distributed throughout the calcite, and that the crystal lattice retains its continuity around the embedded magnetite particles. Characterisation of these calcite/magnetite nanocomposites confirmed their magnetic properties. This new experimental approach is expected to be quite general, such that a small family of block copolymers could be used to drive the incorporation of a wide range of pre-prepared nanoparticles into host crystals, giving intimate mixing of phases with contrasting properties, while limiting nanoparticle aggregation and migration.
Introduction
The ability to create functional inorganic materials through the combination of individual components with contrasting but complementary properties is currently receiving considerable attention due to its promise of materials with novel, tailored properties.1,2 Traditional methods of synthesising inorganic/inorganic composites such as mechanical mixing and annealing generally lead to poor definition of microstructure, and greater control can be achieved using more complex processing such as the formation of alternating layers by physical deposition or chemical solution processing.3 Nanoparticles can also be simply adsorbed or precipitated onto the surface of another crystal,4,5 although this does not generate 3D homogeneity. There is also considerable interest in the incorporation of metal nanoparticles within oxide thin films using methods such as vapour or chemical solution deposition and in situ nanoparticle growth during film formation.6,7 However, synthesis of such materials is often complicated by nanoparticle agglomeration and migration, and nanoparticles are typically located between grain boundaries.7 Methods that offer true nano-scale mixing of distinct inorganic phases are rather limited, but include approaches such as the entrapment of nanoparticles within porous media, e.g. zeolites.8 Alternatively, superlattices have been created by co-assembly of differing nanoparticles, and binary colloidal crystals by layer-by-layer assembly methods.9In this article, we introduce a facile strategy which leads to nanocomposites in which inorganic nanoparticles are uniformly distributed throughout a single crystal matrix with true nano-scale mixing. The method employs functionalised inorganic nanoparticles as simple crystal growth additives, and is based upon prior observations that certain organic additives, ranging from small molecules,10,11 to nanometer-scale micelles,12 sub-micron latex particles13,14 and gels15,16 can sometimes be occluded within single crystals, depending on the structures of the additives and crystal and the solution conditions. However, there is as yet little understanding of the design rules governing such occlusion, which has restricted its application to a limited number of systems employing tailor-made additives. Overcoming this problem, we show here for the first time that highly effective incorporation of nanoparticles within a single crystal host can be achieved by controlling the nanoparticle surface chemistry using a physically adsorbed double hydrophilic diblock copolymer. Such an approach is potentially generic and therefore avoids the need to design and synthesise a bespoke additive for each type of nanoparticle. This methodology can potentially be applied to a huge number of nanoparticle/host crystal systems, where its experimental simplicity makes it an extremely attractive and general method for generating composite materials.
Results
A model system was employed – the incorporation of magnetite nanoparticles within calcite single crystals – to demonstrate the feasibility of forming nanocomposites by using surface-functionalised nanoparticles. Calcite was selected as it has been demonstrated that particles can be occluded within this crystal12,13 while also enabling us to build on previously developed mechanistic understanding. Magnetite nanoparticles, in turn, can be readily distinguished from the CaCO3 matrix, and confer additional physical properties to the parent crystal. Our goal was therefore to establish a proof-of-principle of the methodology rather than to provide a novel method for the synthesis of magnetic CaCO3. Indeed, a number of methodologies have already been described which generate magnetite/CaCO3 composites, but all of these lead to incorporation of magnetite nanoparticles within polycrystals (where particles will inevitably be located between crystallites), rather than within single crystals.2,17–19Design of a suitable block copolymer stabiliser was made based on the requirements for it to adsorb onto the selected inorganic nanoparticles and to the surface of the host crystal. A novel double-hydrophilic diblock copolymer, poly(methacrylic acid)-poly(sodium 4-vinylbenzenesulfonate) (PMAA-PStSNa), was therefore synthesised using reversible addition-fragmentation chain transfer (RAFT) polymerisation.20 Magnetite nanoparticles (MNPs) (Fig. 1) were then precipitated in the presence of this putative steric stabiliser using an established literature protocol.21 Transmission electron microscopy (TEM) analysis showed that the resulting polymer-stabilised MNPs (PS-MNPs) had mean diameters of ≈8 nm (Fig. S1a†), while confirmation of the magnetite polymorph was obtained by powder XRD (Fig. S1b†). Thermogravimetric analysis (TGA) showed that these PS-MNPs comprise ≈50 wt% copolymer, 41 wt% Fe3O4 and 9 wt% water (Fig. S2†). Information on the structure of the polymer-stabilised nanoparticles was obtained from aqueous electrophoresis studies which indicated negative zeta potentials (−30 to −40 mV) from pH 2 to pH 12, suggesting the presence of anionic diblock copolymer chains on the MNP surface (Fig. 2). Moreover, the relatively weak pH-dependence is consistent with the weakly acidic PMAA being adsorbed at the MNP surface as an anchor block, while the strongly acidic PStSNa block acts as a solvated stabiliser block. In contrast, bare magnetite sols exhibited positive zeta potentials from pH 2 to 7, negative zeta potentials from pH 7 to 12 and an isoelectric point at pH ≈ 7.
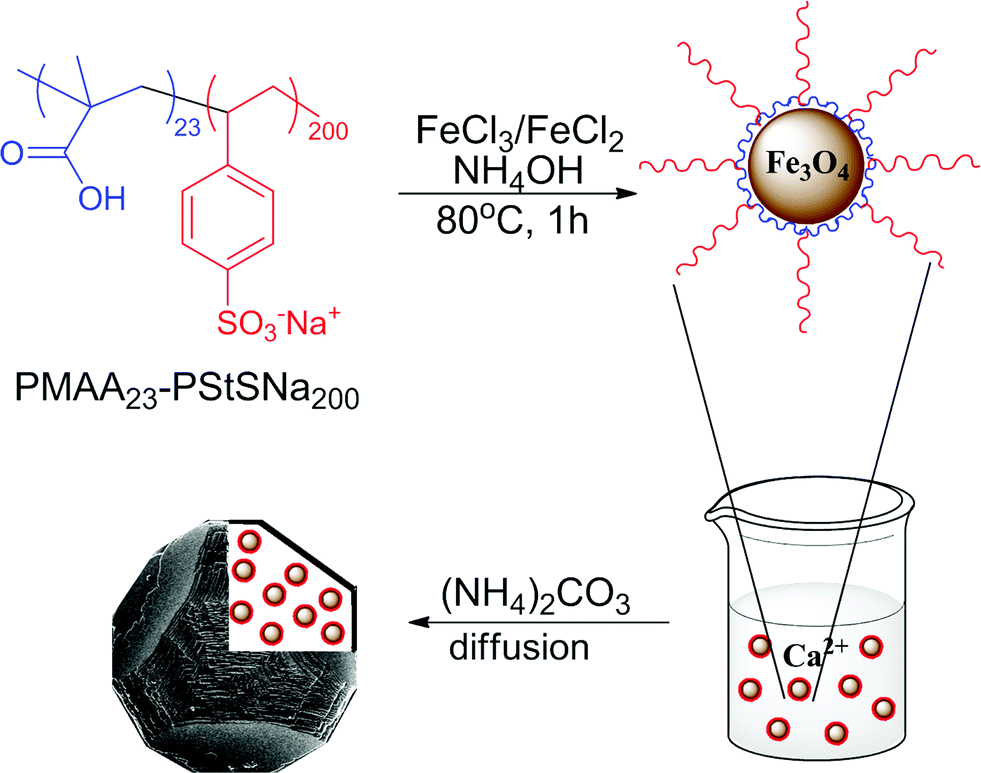 | ||
| Fig. 1 Schematic representation of the synthesis of magnetite nanoparticles stabilised with the diblock copolymer PMAA23-PStSNa200, and their occlusion within calcite single crystals. | ||
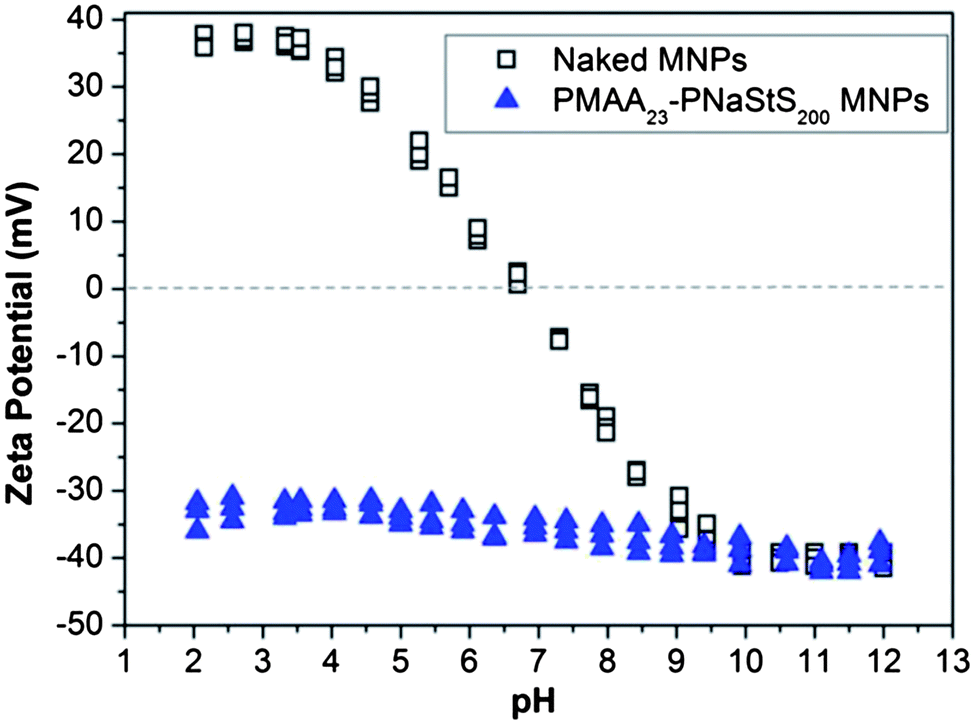 | ||
Fig. 2 Aqueous electrophoresis data obtained for magnetite sols coated with PMAA23-PStSNa200 ( ); no copolymer ( ); no copolymer ( ). Electrophoretic measurements were made on 0.01 wt% dispersions in the presence of 10−3 M background NaCl. ). Electrophoretic measurements were made on 0.01 wt% dispersions in the presence of 10−3 M background NaCl. | ||
Calcite single crystal/magnetite nanoparticle nanocomposites were then prepared by a simple one-pot method in which CaCO3 was precipitated in the presence of the functionalised MNPs using the ammonia diffusion method.22 Control experiments were also performed in which CaCO3 was precipitated in additive-free solution or in the presence of bare MNPs at Ca2+ concentrations ranging from 1.5 mM to 20 mM, and rhombohedral calcite crystals were produced in all cases (Fig. S3†). As the goal of our synthesis was to occlude the magnetic nanoparticles within single crystals rather than within polycrystalline structures, the reaction conditions were varied to identify optimal synthesis conditions. Overall, a transition from single crystals to polycrystalline particles was observed with increasing [Ca2+] and PS-MNP concentrations, while reduced yields were obtained at lower calcium and PS-MNP concentrations (summarised in Fig. S4 and Table S1†). Thus, rhombohedral single crystals of calcite were produced at low reagent concentrations (1.5 mM Ca2+ and 0.1 mg ml−1 PS-MNPs), while increasing the [Ca2+] at a constant [PS-MNP] of 0.10 mg ml−1 generated inter-grown calcite crystals and polycrystalline calcite particles at [Ca2+] = 10 mM and [Ca2+] = 20 mM, respectively. When the PS-MNPs were present at higher concentrations of 0.4–4.0 mg ml−1, the transition to polycrystalline particles occurred at lower Ca2+ concentrations (6–10 mM).
A balance between the yield of nanocomposite crystals and maintenance of single crystal structure was achieved when [Ca2+] = 3 mM and [PS-MNP] = 4.0 mg ml−1, so crystals produced under these conditions were selected for further detailed analysis. These crystals exhibited morphologies that are characteristic of “mesocrystals” precipitated in the presence of poly(sodium 4-styrenesulfonate)23 or structurally similar copolymers.24 These displayed a combination of smooth and rough faces, and truncated edges and were typically [001]-oriented (Fig. 3). That the coated MNPs were occluded within the calcite crystals was immediately demonstrated by their appearance and magnetic properties. While calcite crystals precipitated in the absence of PS-MNPs or in the presence of uncoated MNPs were invariably white, those prepared in the presence of the PS-MNPs were yellow-grey in colour and responded to an external magnetic field. Occlusion was confirmed and quantified using atomic absorption spectroscopy (AAS). This technique indicated that the nanocomposites comprised approximately 5.4 wt% of Fe3O4, which corresponds to ∼12.5 wt% of PS-MNPs.
 | ||
| Fig. 3 SEM images of calcite crystals precipitated after 1 day from solutions containing [Ca2+] = 3.0 mM and 4.0 mg ml−1 PS-MNP. | ||
The spatial distribution of the MNPs throughout the calcite phase was investigated using SEM and TEM. Examination of the cross-section through a fractured crystal using SEM indicated a uniform distribution of PS-MNPs throughout the crystal (Fig. 4a), while EDX confirmed the presence of Fe within the crystal (Fig. S5a†). Specimens for TEM analysis were prepared using Focused Ion Beam (FIB) where sections from three different crystals were successfully prepared and examined. A representative TEM image is presented in Fig. 4b, while the entire section and its position in the original crystal are shown in Fig. S6.† Both high resolution TEM (HR-TEM) imaging (Fig. 4c) and selected area electron diffraction (SAED) (Fig. 4d) of all sections confirmed that the calcite phase was a single crystal, where patterns were recorded with a 300 nm aperture. These images demonstrated that the magnetite nanoparticles were homogeneously distributed throughout the calcite, with no evidence of aggregation, and that there was no discontinuity with the host calcite lattice. Electron diffraction also confirmed the presence of magnetite within the calcite crystal, where powder rings corresponding to the magnetite nanoparticles were observed when the sample was tilted to reduce diffraction from the calcite crystal itself (Fig. 4e and at higher magnification in Fig. S5b†).
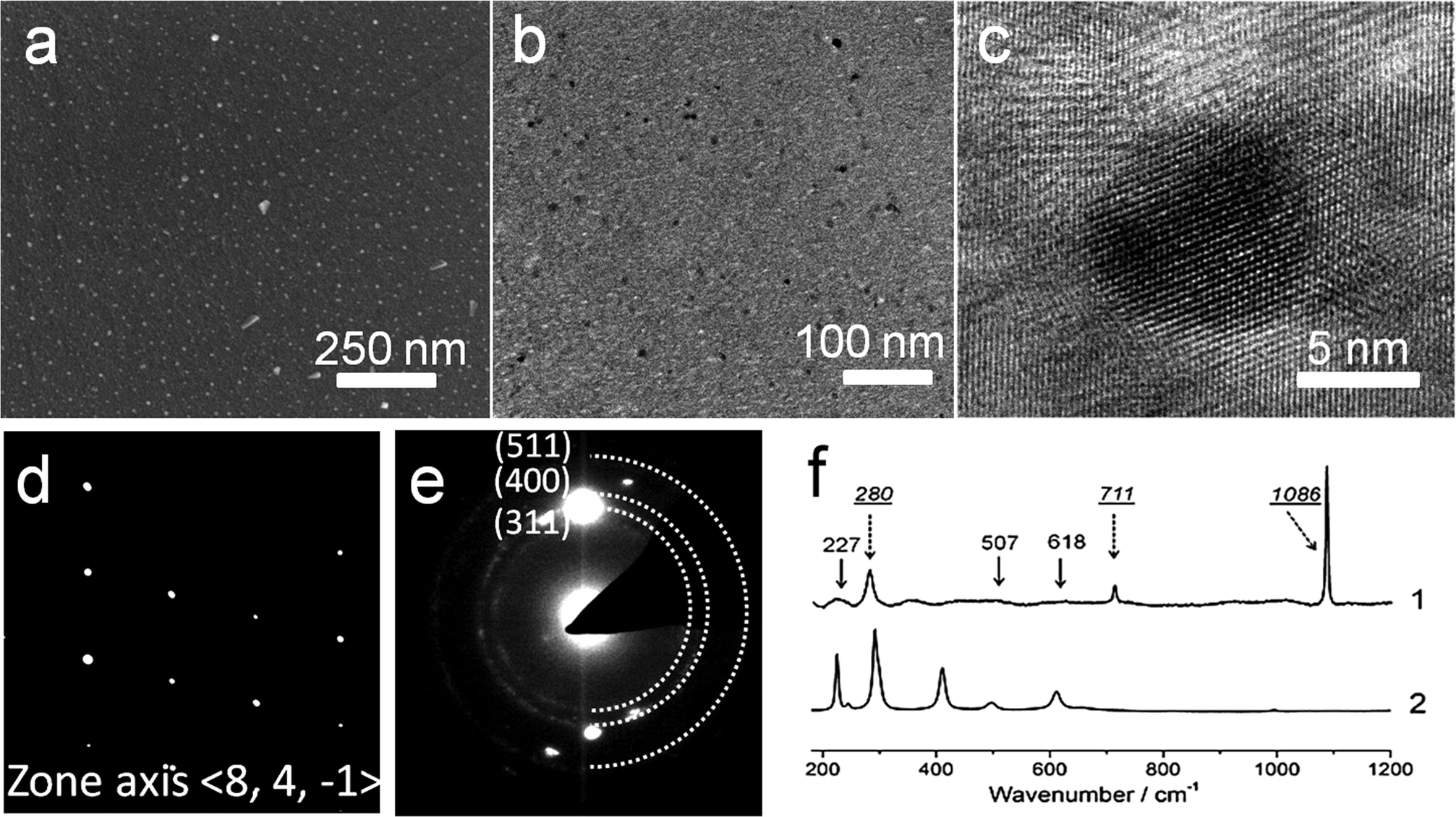 | ||
| Fig. 4 (a) SEM image of the cross-section through a calcite single crystal prepared in the presence of PS-MNPs. (b) TEM image of a thin section through the nanocomposite crystals, and (c) a high resolution TEM image showing the continuity of the crystal lattice around an embedded magnetite particle. (d) An electron diffraction pattern of the same sample, showing that it is a single crystal of calcite, and (e) an electron diffraction pattern, obtained by tilting the crystal off the diffraction angle of calcite, showing rings corresponding to magnetite. (f) Raman spectra of (1) nanocomposite crystals after annealing at 500 °C to remove the copolymer and (2) PS-MNPs after annealing at 500 °C. | ||
These nanocomposite crystals were also characterised using Raman microscopy and TGA. As the copolymer coating the MNPs fluoresces when excited by the Raman laser, spectra clearly showing the mineral phases could only be obtained after annealing the nanocomposite particles at 500 °C to pyrolyse the copolymer. This thermal treatment is accompanied by the conversion of magnetite to hematite. The annealed particles showed characteristic calcite bands at 155 and 282 (lattice modes), 710 (ν4) and 1085 cm−1 (ν1),22 together with bands at 227 (A1g), 507 (A1g) and 618 (Eg) cm−1 due to hematite (Fig. 4f).25 The shoulder starting to appear at around at 200 cm−1 in spectrum 1 belongs to the calcite lattice mode at 155 cm−1. TGA yielded data consistent with nanoparticle occlusion within the calcite, but quantitative analysis of the heating curves was difficult as the CaCO3, Fe3O4 and diblock copolymer components of the nanocomposites all undergo mass loss on heating over similar temperature ranges (Fig. S7†).
The magnetic properties of these nanocomposite crystals and the PS-MNPs were investigated using vibrating sample magnetometry from 4 K to 293 K with a sensitivity of around 5 emu. Both the nanocomposite crystals and the PS-MNPs are superparamagnetic (i.e. single domain crystals) at room temperature, but become ferrimagnetic at low temperatures. Magnetisation values were determined at 5 K using an applied magnetic field of 5 T, and were recorded as 11 emu g−1 of polymer-stabilised MNPs (which is equivalent to 27.5 emu g−1 of magnetite) and 1.8 emu g−1 for the calcite nanocomposite samples (Fig. 5a). This shows that the composite crystals contain approximately 6.5 wt% of magnetite or 15.8 wt% PS-MNPs, which is in good agreement with the values determined by atomic absorption.
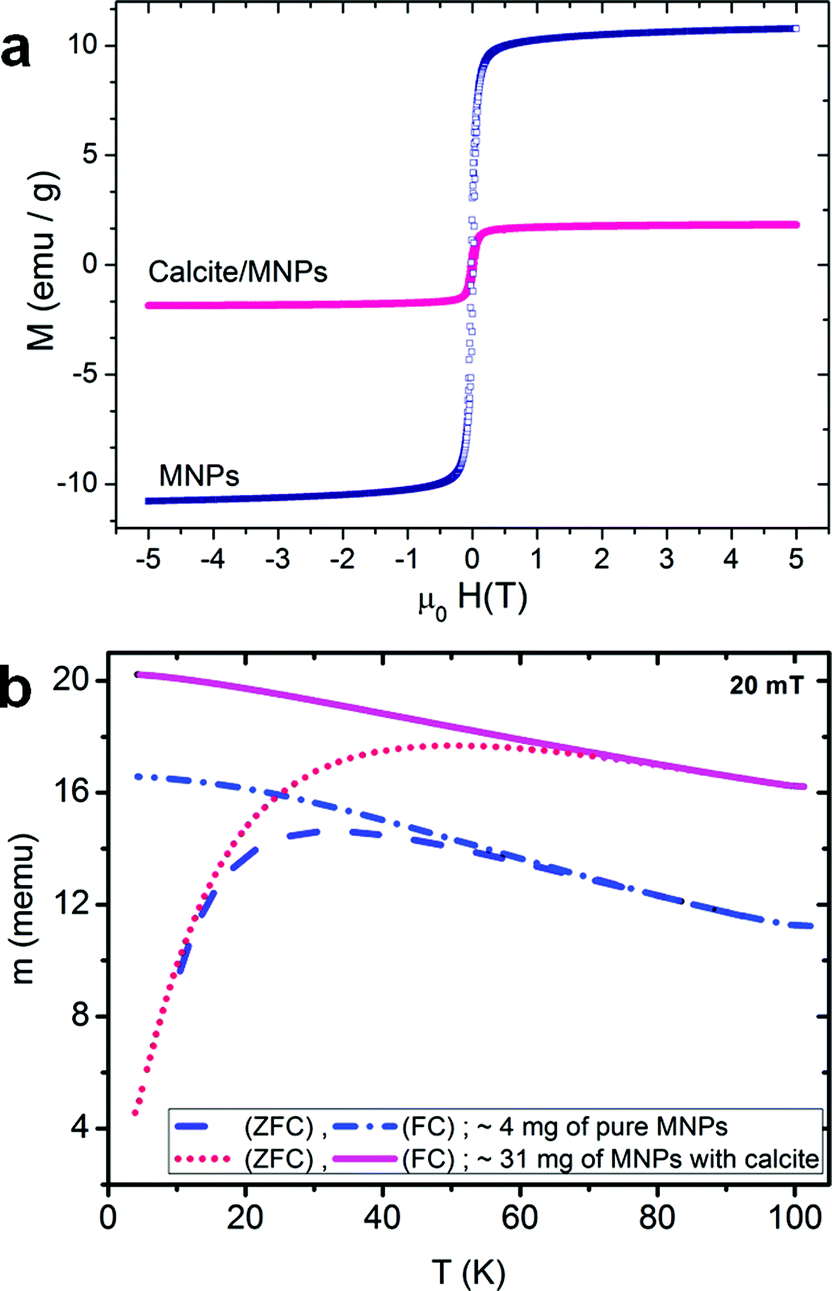 | ||
| Fig. 5 (a) Magnetisation loops measured at 5 K, and (b) thermomagnetic zero-field-cooled/field-cooled (ZFC/FC) curves of calcite crystals prepared in the presence of PS-MNPs and of these copolymer-coated MNPs alone. | ||
These magnetisation values are quite low compared with the saturation magnetisation of ∼92 emu g−1 for bulk magnetite.26 Although the magnetisation of magnetite is size-dependent, where spherical 5 nm Fe3O4 nanoparticles has previously been measured at ≈50 emu g−1 at 5 K,27 the relatively low value recorded here suggests that the magnetite nanoparticles are poorly crystalline, as is often observed for synthesis in the presence of polymers; they may also exhibit an Fe2O3 surface layer which can cause magnetisation changes.28 This hypothesis is also consistent with the observation that powder XRD spectra of the composite crystals failed to show clear spectra corresponding to magnetite. Both samples have a coercive field of 20 mT at 5 K and a remnant to 5 T magnetisation ratio of 0.25, while the blocking temperatures for the PS-MNPs and the calcite sample are 32 K and 48 K respectively, which is consistent with an average particle size of about 5 nm for the active magnetic component (Fig. 5b).27 The difference in the blocking temperature can be attributed to small deviations in particle size (5–6 nm), or to the different particle environment which causes small but significant changes in the magnetic anisotropy.28 Given that TEM shows that the PS-MNPs are well-dispersed within the calcite lattice and that the coercivity and remnance are the same for both the nanocomposite crystals and the PS-MNPs, it is likely that a change in the surface states of the PS-MNPs leads to greater anisotropy.28
Discussion
Although crystallisation is widely used as a purification method, with impurities being expelled from the growing crystal lattice, it is now recognised that appropriate reaction conditions can lead to retention of impurities within single crystals. For example, a rigid matrix can be readily engulfed by a growing crystal, as occurs during templating processes,29–31 while occlusion of a softer gel matrix depends on the balance between gel stiffness and the rate of crystal growth.15 The entrapment of fully mobile, soluble additives present in the crystal growth solution is clearly far more demanding, but the formation of single crystal biominerals occluding biomacromolecules,32 and the encapsulation of dyes11 and small molecules10 provides some precedent for the approach. This strategy has also been extended to the encapsulation of sub-micron polymer latexes within ZnO14 and calcium carbonate,13 and more recently to the encapsulation of 20 nm diblock copolymer micelles within calcite crystals.12Considering potential mechanisms for particle occlusion within a crystal, the formation of micron- to millimetre-sized solution, gas and particle inclusions within crystals is well known,33 and a substantial body of literature covers theories of particle encapsulation during melt growth.34,35 For particles of these sizes, occlusion only occurs above a certain critical rate of crystallisation, which depends on the particle size, solid–liquid interfacial energy, van der Waals interactions and density changes caused by crystallisation.36 Above this critical rate, the crystallisation front effectively overtakes the impurities, leading to engulfment.37 At the other end of the size regime, the occlusion of nanoscale impurities within solution-phase crystal growth depends on competition between the incoming crystal growth units and the impurity at the kinks and steps which constitute the active growth sites.33,38 Both the strength of adsorption of the impurity particles to these sites and the crystal growth kinetics therefore govern particle occlusion. Thus, at a fixed concentration of strongly adsorbing impurity, the concentration of particles on kink sites and steps will decrease with increasing supersaturation, due to increasing competition with host units, leading to reduced incorporation. Weakly adsorbed particles, in contrast, are expected to show superior incorporation with increasing supersaturation, as the probability of preservation at the growth sites is the dominant factor for occlusion of these particles.
The results presented here demonstrate that this basic understanding of the mechanisms underlying the incorporation of additive species within single crystals can be readily used for the rational design of appropriate double-hydrophilic diblock copolymers which can be used to achieve efficient occlusion of inorganic nanoparticles within a host single crystal. Sulphonate groups are well-known to bind strongly to CaCO3,24 and strong acid/weak acid diblock copolymers were therefore synthesised which could bind strongly both to the inorganic particles (via the carboxylic acid-based PMAA block) and the host crystal (via the sulphonate-based PNaStS block). In designing such copolymers, previous work had also suggested that particles functionalised by anionic polyelectrolyte stabiliser chains are far more effectively incorporated than similarly charged “hard sphere” particles.13 Finally, the polymer coating also plays a key role by conferring colloidal stability on the nanoparticles in the crystal growth solution, where this is difficult to achieve with say, nanoparticles coated with anionic ω-functionalised thiols (unpublished data). By adopting this method, it is therefore no longer necessary to redesign and synthesise from scratch every particle which one wishes to occlude within a crystal. The same polymer can be used to drive the occlusion of a wide range of nanoparticles within single crystal hosts, making this a very general approach to create composite materials with controlled nanostructures.39
Conclusions
In summary, we have described a facile one-pot method which leads to the occlusion of inorganic nanoparticles within a single crystal matrix. The nanoparticles are employed as crystal growth additives, where tailoring of their surfaces with a physically adsorbed diblock copolymer enables their effective incorporation within the host crystal. In this context, the copolymer plays two essential roles; it confers colloidal stability in the crystal growth solution, and it ensures that the nanoparticles bind strongly to the growing crystal surface, thus facilitating their occlusion. This strategy provides a unique way of achieving nano-scale mixing of the separate component phases, while minimising problems of nanoparticle agglomeration and migration. Although this paper focused on magnetite nanoparticles in calcite single crystals as a convenient model system, it is envisaged that this approach will be quite general. Appropriate choice of diblock copolymer stabilisers can enable occlusion of a wide range of “guest” nanoparticles within single crystal matrices, thereby endowing additional functionality on the host crystal, and providing the potential for new, emergent properties.Acknowledgements
We thank the EPSRC for financial support via grants EP/G00868X/1 (AK, MS, FCM and SPA) and EP/K006304/1 (AK, FCM, LAF and SPA). This work was also supported by an EPSRC Leadership Fellowship (EP/H005374/1; FCM, YYK and JI) and an ERC Advanced Investigator grant (PISA 320372; SPA).Notes and references
- G. S. Rohrer, M. Affatigato, M. Backhaus, R. K. Bordia, H. M. Chan, S. Curtarolo, A. Demkov, J. N. Eckstein, K. T. Faber, J. E. Garay, Y. Gogotsi, L. P. Huang, L. E. Jones, S. V. Kalinin, R. J. Lad, C. G. Levi, J. Levy, J. P. Maria, L. Mattos, A. Navrotsky, N. Orlovskaya, C. Pantano, J. F. Stebbins, T. S. Sudarshan, T. Tani and K. S. Weil, J. Am. Ceram. Soc., 2012, 95, 3699–3712 CrossRef CAS.
- D. Walsh, Y. Y. Kim, A. Miyamoto and F. C. Meldrum, Small, 2011, 7, 2168–2172 CrossRef CAS PubMed.
- C. W. Nan, M. I. Bichurin, S. X. Dong, D. Viehland and G. Srinivasan, J. Appl. Phys., 2008, 103, 35 CrossRef.
- G. Evans, G. V. Duong, M. J. Ingleson, Z. L. Xu, J. T. A. Jones, Y. Z. Khimyak, J. B. Claridge and M. J. Rosseinsky, Adv. Funct. Mater., 2010, 20, 231–238 CrossRef CAS.
- X. Y. Chu, X. Hong, X. T. Zhang, P. Zou and Y. C. Liu, J. Phys. Chem. C, 2008, 112, 15980–15984 CAS.
- G. Walters and I. P. Parkin, J. Mater. Chem., 2009, 19, 574–590 RSC.
- N. Bahlawane, K. Kohse-Hoinghaus, T. Weimann, P. Hinze, S. Rohe and M. Baumer, Angew. Chem., Int. Ed., 2011, 50, 9957–9960 CrossRef CAS PubMed.
- J. Dong, Z. H. Xu and S. M. Kuznicki, Adv. Funct. Mater., 2009, 19, 1268–1275 CrossRef CAS.
- K. P. Velikov, C. G. Christova, R. P. A. Dullens and A. van Blaaderen, Science, 2002, 296, 106–109 CrossRef CAS PubMed.
- S. Borukhin, L. Bloch, T. Radlauer, A. R. Hill, A. H. Fitch and B. Pokroy, Adv. Funct. Mater., 2012, 22, 2416–2422 CrossRef.
- B. Kahr and R. W. Gurney, Chem. Rev., 2001, 101, 893–951 CrossRef CAS PubMed.
- Y. Y. Kim, K. Ganesan, P. Yang, A. N. Kulak, S. Borukhin, S. Pechook, L. Ribeiro, R. Kröger, S. J. Eichhorn, S. P. Armes, B. Pokroy and F. C. Meldrum, Nat. Mater., 2011, 10, 890–896 CrossRef CAS PubMed.
- Y. Y. Kim, L. Ribeiro, F. Maillot, O. Ward, S. J. Eichhorn and F. C. Meldrum, Adv. Mater., 2010, 22, 2082–2086 CrossRef CAS PubMed.
- R. Munoz-Espi, G. Jeschke, I. Lieberwirth, C. M. Gomez and G. Wegner, J. Phys. Chem. B, 2007, 111, 697–707 CrossRef CAS PubMed.
- H. Y. Li and L. A. Estroff, Adv. Mater., 2009, 21, 470–473 CrossRef CAS.
- H. Y. Li, H. L. Xin, D. A. Muller and L. A. Estroff, Science, 2009, 326, 1244–1247 CrossRef CAS PubMed.
- R. F. Fakhrullin, A. G. Bikmullin and D. K. Nurgaliev, ACS Appl. Mater. Interfaces, 2009, 1, 1847–1851 CAS.
- Y. Zhao, Y. Lu, Y. Hu, J.-P. Li, L. Dong, L. Lin and S.-H. Yu, Small, 2010, 6, 2436–2442 CrossRef CAS PubMed.
- M. Mihai, V. Socoliuc, F. Doroftei, E.-L. Ursu, M. Aflori, L. Vekas and B. C. Simionescu, Crystal Growth and Design, 2013, 13, 3535–3545 CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- J. J. Yuan, S. P. Armes, Y. Takabayashi, K. Prassides, C. A. P. Leite, F. Galembeck and A. L. Lewis, Langmuir, 2006, 22, 10989–10993 CrossRef CAS PubMed.
- J. Ihli, P. Bots, A. Kulak, L. G. Benning and F. C. Meldrum, Adv. Funct. Mater., 2013, 23, 1965–1973 CrossRef CAS.
- R. Q. Song and H. Colfen, Adv. Mater., 2010, 22, 1301–1330 CrossRef CAS PubMed.
- A. N. Kulak, P. Iddon, Y. T. Li, S. P. Armes, H. Colfen, O. Paris, R. M. Wilson and F. C. Meldrum, J. Am. Chem. Soc., 2007, 129, 3729–3736 CrossRef CAS PubMed.
- A. M. Jubb and H. C. Allen, ACS Appl. Mater. Interfaces, 2010, 2, 2804–2812 CAS.
- J. Choi, J. Cha and J. K. Lee, RSC Adv., 2013, 3, 8365–8371 RSC.
- G. F. Goya, T. S. Berquo, F. C. Fonseca and M. P. Morales, J. Appl. Phys., 2003, 94, 3520–3528 CrossRef CAS.
- A. Tamion, C. Raufast, M. Hillenkamp, E. Bonet, J. Jouanguy, B. Canut, E. Bernstein, O. Boisron, W. Wernsdorfer and V. Dupuis, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 144403 CrossRef.
- B. Wucher, W. B. Yue, A. N. Kulak and F. C. Meldrum, Chem. Mater., 2007, 19, 1111–1119 CrossRef CAS.
- W. B. Yue, A. N. Kulak and F. C. Meldrum, J. Mater. Chem., 2006, 16, 408–416 RSC.
- A. S. Finnemore, M. R. J. Scherer, R. Langford, S. Mahajan, S. Ludwigs, F. C. Meldrum and U. Steiner, Adv. Mater., 2009, 21, 3928–3932 CrossRef CAS.
- H. A. Lowenstam and S. Weiner, On Biomineralization, Oxford University Press, New York, 1989 Search PubMed.
- A. A. Chernov, in Springer Series in Solid State Sciences, Springer-Verlag, 1984 Search PubMed.
- D. Coupard, F. Girot and J. M. Quenisset, J. Mater. Sci., 1996, 31, 5305–5308 CrossRef CAS.
- A. W. Rempel and M. G. Worster, J. Cryst. Growth, 1999, 205, 427–440 CrossRef CAS.
- M. S. Park, A. A. Golovin and S. H. Davis, J. Fluid Mech., 2006, 560, 415–436 CrossRef.
- D. R. Uhlmann, B. Chalmers and K. A. Jackson, J. Appl. Phys., 1964, 35, 2986–2993 CrossRef CAS.
- L. Stappers and J. Fransaer, J. Electrochem. Soc., 2006, 153, C472–C482 CrossRef CAS PubMed.
- A. N. Kulak, P. Yang, Y-Y. Kim, S. P. Armes and F. C. Meldrum, Chem. Commun., 2013 10.1039/C3CC47904H.
Footnote |
| † Electronic supplementary information (ESI) available: Full description of experimental methods and further characterisation data. See DOI: 10.1039/c3sc52615a |
| This journal is © The Royal Society of Chemistry 2014 |