 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUtilizing the bioorthogonal base-pairing system of L-DNA to design ideal DNA nanocarriers for enhanced delivery of nucleic acid cargos†
Kyoung-Ran
Kim
ad,
Taemin
Lee
c,
Byeong-Su
Kim
c and
Dae-Ro
Ahn
*ab
aCenter for Theragnosis, Biomedical Research Institute, Korea Institute of Science and Technology, Hwarangno 14-gil 5, Seongbuk-gu, Seoul 136-791, Republic of Korea. E-mail: drahn@kist.re.kr; Fax: +82 2 958 5909; Tel: +82 2 958 6645
bKIST campus, University of Science and Technology (UST-KIST), Hwarangno 14-gil 5, Seongbuk-gu, Seoul 136-791, Republic of Korea
cInterdisciplinary School of Green Energy, Ulsan National Institute of Science and Engineering (UNIST), Ulsan 689-798, Republic of Korea
dDepartment of Chemistry, College of Science, Yonsei University, Republic of Korea
First published on 6th January 2014
Abstract
DNA nanoconstructs are a potential drug carrier with high biocompatibility. They are promising particularly when therapeutic nucleic acids are the cargos to be delivered, since both the carrier and the cargo are nucleic acids which can be designed, synthesized and assembled in a seamless feature without using post-synthetic conjugation chemistry. However, the unwanted base-paring events between the cargo and the carrier may potentially disturb the desired structure of the cargo-loaded carrier. To address this concern, we propose a DNA nanocarrier composed of L-DNA strands having a bioorthogonal base-paring system. The study presented here provides useful properties of L-DNA as a backbone for the DNA nanocarrier and demonstrates superiority of L-DNA over the natural D-DNA backbone in the delivery of an anti-proliferative aptamer as well as in the construction of the cargo-carrier assembly.
DNA nanocarriers are an emerging class in drug delivery technologies.1,2 Unlike most of polymer-based carriers, they are fully biodegradable into non-toxic small nucleotides and easily controllable in size and shape at a tailor-made level.3 They are often internalized in mammalian cells in the absence of any transfection agent.4,5 These properties are valuable when they are employed in biomedical applications. Recently, DNA nanocarriers have been utilized in intracellular delivery of bioactive molecules such as anticancer drugs,1,2 aptamers,6 antisenses,7 immunogenic molecules,8–10 and siRNA.11,12 DNA nanocarriers are particularly useful in delivery of nucleic acid cargos, since the vehicle and the cargo are the same nucleic acids and can thus be designed and prepared in an integrated feature without any additional conjugation chemistry. For the successful assembly of a cargo-loaded carrier, however, the sequences of the carrier and the cargo should be carefully designed and selected since not only the DNA nanocarrier but the oligonucleotide-based cargo also employs the A–T and G–C base-paring patterns for construction of active motifs, which has a potential to disturb the self-assembly of the DNA nanocarrier. The probability of undesired assembly resulting from this interference would increase when more than one kind of nucleic acid-based cargos are attached to the carrier. In order to address this issue and to freely choose cargo molecules regardless of the sequence of nanocarriers, the carriers are required to have a base-paring system that is orthogonal to the natural base-pairing system. In addition, the orthogonal base-paring system having thermodynamic properties identical to the natural system would be preferred for forming the consistent DNA nanostructure even after replacing the backbone of the carriers with the orthogonal system.
Although several modified nucleic acids have been reported to have the orthogonal base-paring property up to now, few cases show similar thermodynamic stability as that of natural DNA.13–16L-DNA, the mirror form of natural D-DNA has thermodynamically identical properties in hybridization (Fig. 1a).17–19 Therefore, if L-DNA instead of D-DNA is employed as a backbone for the assembly of the nanocarriers, any natural nucleic acid-based cargo could be used without concerns about interference from undesired hybridization between the cargo sequence and the carrier sequence. To test this concept in this study, we prepared L-DNA tetrahedron (L-Td) in which a therapeutic, anti-proliferative aptamer is dangled at vertices of the tetrahedron. We analysed the biophysical properties of this L-Td-based multivalent aptamer and evaluated its anti-proliferative potency in cancer cells in comparison with the D-Td-based one.
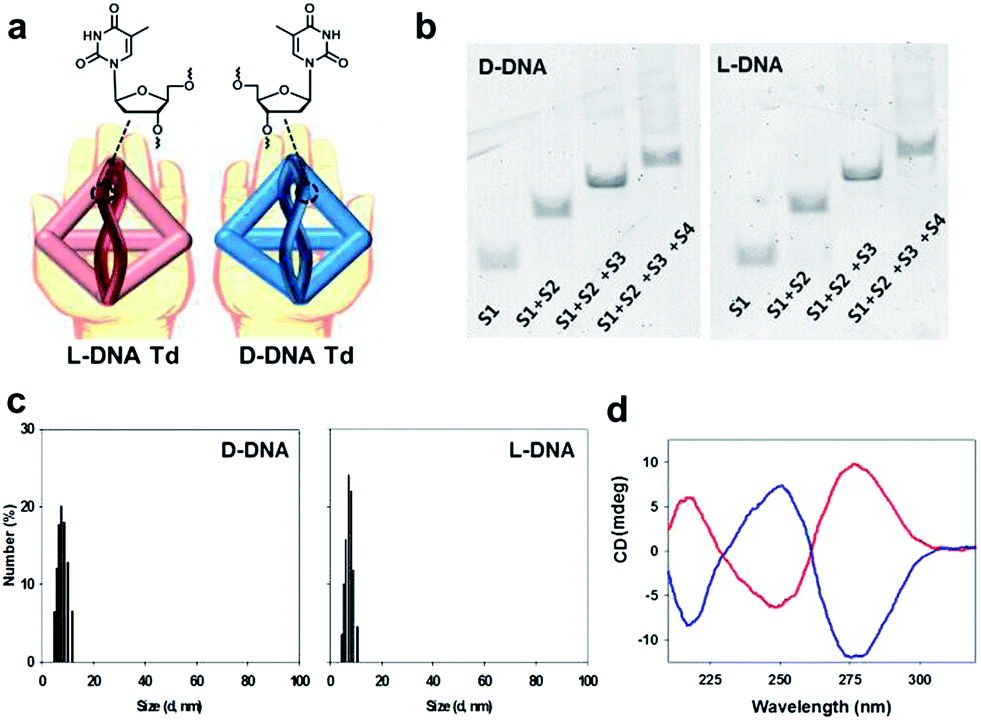 | ||
| Fig. 1 (a) Schematic illustration for D-Td and L-Td. (b) Native PAGE to verify assembly of D-Td (left) and L-Td (right). (c) DLS data of D-Td (left) and L-Td (right). (d) CD spectra of D-Td (blue) and L-Td (red). | ||
The DNA oligonucleotides required for the construction of Tds were synthesized by using the standard protocols in a DNA synthesizer. The base sequences of DNA strands were adopted from Tuberfield's Td (ESI, Table S1†).20 The Td assembly without aptamer attachment was performed by following the previously reported procedure.20 The construction of self-assembled D- and L-Td was verified on 6% non-denaturing polyacrylamide gel electrophoresis (PAGE) (Fig. 1b). Both D- and L-Td showed the same sequential retardation pattern as the number of DNA strands participating in the assembly increased, suggesting that L-Td could be formed under the identical condition required for the assembly of D-Td. Hydrodynamic sizes of D- and L-Td were 7.63 (±0.37) nm and 7.80 (±0.31) nm, respectively (Fig. 1c), as determined by dynamic light scattering (DLS). The DNA nanostructures were also characterized by atomic force microscopy (AFM) images showing about 3.79 (±0.56) nm of height for D-Td and 3.83 (±0.51) nm of height for L-Td at the dried state, respectively (ESI, Fig. S1†). When CD spectra of Tds were measured, a typical profile for a right-handed DNA helix was observed from D-Td presenting a negative peak around 254 nm and a positive peak around 270 nm, whereas the symmetrical mirror spectrum was observed from L-Td, indicating that L-Td was formed with the left-handed helical property as expected (Fig. 1d).
It has been reported that D-Td could be internalized into mammalian cells via endocytosis.1,4 In order to investigate the influence of the backbone on this cellular uptake property of Td, cellular uptake efficiency of L-Td was also examined and compared with that of D-Td. After incubation of fluorescein-labelled Tds either with cancer cells (HeLa) or with normal cells (NIH-3T3) in the absence of any transfection reagent, fluorescence microscopic images were obtained. Fluorescence intensity in the cells treated with L-Td was comparable to that observed with D-Td, which suggests that L-Td could be delivered as efficiently as D-Td (Fig. 2a). Quantitative analysis based on flow cytometry showed that intracellular delivery of L-Td was about 2.5-fold higher in the cancer cell line and 3-fold higher in the normal cell line than the internalization of D-Td, respectively (Fig. 2b). Previously, we have reported that non-clathrin-mediated endocytosis mechanisms were involved with the uptake of D-Td.1 A test using three endocytosis inhibitors to verify the delivery mechanism for L-Td revealed that the enhanced uptake of L-Td compared to D-Td was achieved through mechanisms including not only non-clathrin-mediated but also clathrin-mediated endocytosis (Fig. 2c). According to the microscopic data obtained with staining reagents for lysosome, mitochondria and nucleus (ESI, Fig. S2a–c†), the DNA tetrahedron is found in cytoplasm containing lysosome and mitochondria, but not in the nucleus. In addition, the cell fate was not significantly affected by the delivery of the DNA constructs, shown by cell cycle analysis (ESI, Fig. S2d†).
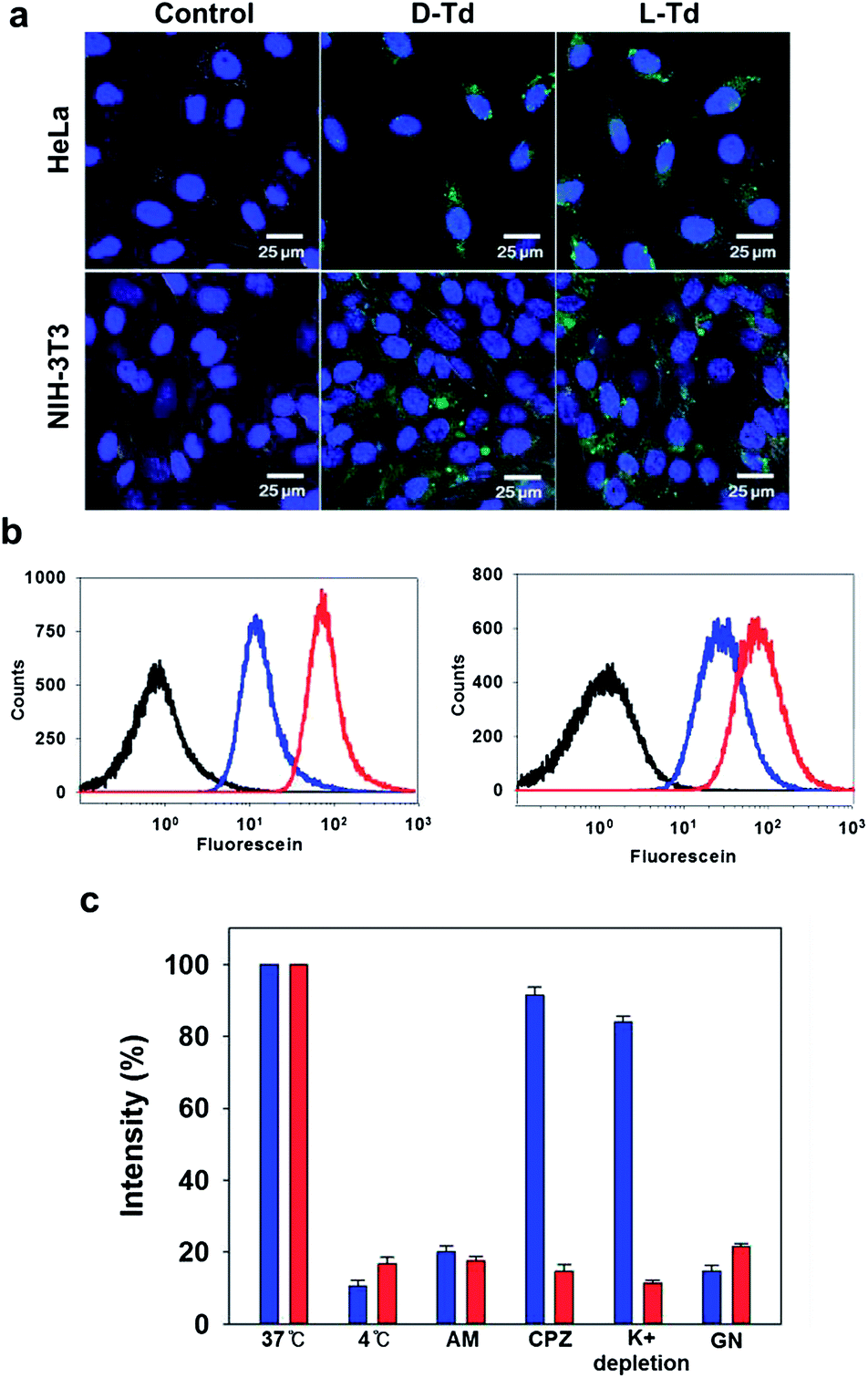 | ||
| Fig. 2 (a) Fluorescence microscopic images of cancer cells (HeLa, top) and normal cells (NIH-3T3, bottom) treated with D-Td (middle, green) and L-Td (right, green). The nuclei were stained with Hoechst34580 (blue). (b) Flow cytometry analysis for the cellular uptake of D-Td (blue) and L-Td (red) into HeLa (left) and NIH-3T3 cells (right). The black traces represent untreated cells. (c) Average cellular fluorescence intensity of D-Td (blue) and L-Td (red) in HeLa cell lysates from the cells cultured in the presence of different endocytosis inhibitors: amiloride (AM, macropinocytosis), chlorpromazine (CPZ, clathrin-mediated), K+ depletion (clathrin-mediated) and genistein (GN, caveolae-mediated). The fluorescence intensity was normalized to the total amount of cellular proteins. | ||
The effect of the mirror backbone on nuclease resistance was also examined in 10% mouse serum. In contrast to D-Td which could hardly survive after 10 h exposure to the serum solution, L-Td was conserved without degradation by nucleases under the same condition, showing invulnerability against the enzymes in serum for several hours (ESI, Fig. S3†). Increased nuclease resistance gained with the Td assembly seemed to be additionally improved due to the unnatural sugar backbone in L-Td.17 This long term serum stability of the L-DNA backbone would potentially be advantageous when it is used as a carrier in intravenous delivery for in vivo applications. When the intracellular stability of the DNA constructs was examined by measuring FRET efficiency of dually labelled DNA Tds in cells (ESI, Fig. S4†), L-Td was far more stable than D-Td, as expected from the serum stability.
After observing useful properties of L-Td as a delivery carrier in comparison with D-Td such as enhanced cellular uptake and higher serum stability, we attempted to assemble aptamer-attached Tds by adopting the DNA strands containing the sequence of an anti-proliferative aptamer, AS1411 at one end (Fig. 3a and ESI, Table S1†).21 Based on the same manner used for construction of D- and L-Td, preparation of DNA tetrahedra containing one to three aptamers were pursued. According to the gel electrophoresis analysis, various bands including non-specifically assembled structures were observed with the D-Td backbone (Ap1-D-Td, Ap2-D-Td and Ap3-D-Td), suggesting that the D-Td construct was not created properly with the aptamer-labeled DNA strands (Fig. 3b, left). Aptamer constructs with L-DNA strands (Ap1-L-Td, Ap2-L-Td and Ap3-L-Td), however, led to clear bands with retarded mobility compared to the band of L-Td itself, implying that the self-assembled Td structures were maintained even with the dangling aptamers (Fig. 3b, right). In addition, D-DNA strands without the aptamer moiety failed to make the self-assembled nanoconstruct exclusively just in the presence of free 24-mer aptamer strands, which did not affect the formation of L-Td (ESI, Fig. S5†). These results demonstrate that the unsuccessful assembly of Ap-D-Tds resulted from the undesired base-pairing between the aptamer moiety and the sequence required for the tetrahedron structure. They also implicated that the cross-interaction based on partial base complementarity between the aptamer and the D-Td skeleton do not exist in the L-Td case, because of the backbone orthogonality. In contrast to the size similarity between D-Td and L-Td, the sizes of Ap3-D-Td (10.9 ± 0.44 nm) and Ap3-L-Td (8.93 ± 0.21 nm) were different from each other in DLS measurement (ESI, Fig. S6†). The size increase in Ap3-D-Td might be due to the failure to construct compactly self-assembled structures, consistent with the gel analysis data.
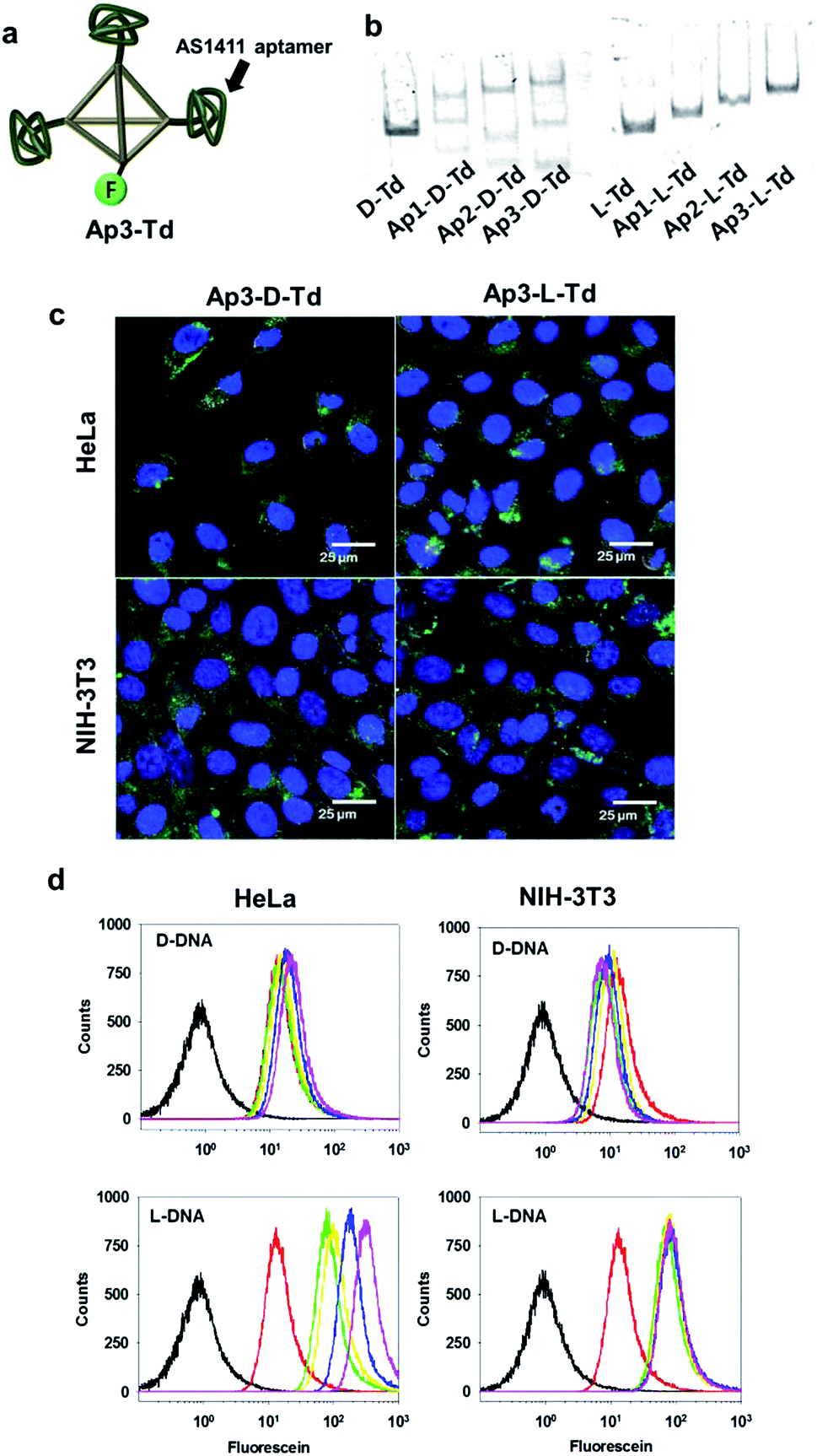 | ||
| Fig. 3 (a) Schematic presentation of Td with three AS1411 aptamers attached. The fluorescein label was denoted by ‘F’. (b) Native PAGE to analyse the assembly of aptamer-labeled Tds. (c) Cellular uptake of aptamer-labeled Tds into both cancer and normal cells (green). The nuclei were stained with Hoechst34580 (blue). (d) Flow cytometry analysis for the cellular uptake of Td (green), Ap1-Td (yellow), Ap2-Td (blue), Ap3-Td (magenta) and free AS1411 aptamer (red) into the cells. The black traces represent the untreated control cells. | ||
To investigate whether organization of the aptamers on Tds influences on the cellular uptake of the DNA nanoconstructs, microscopic images and flow cytometric data of both cancer cells and normal cells were acquired after treatment of the cells with the aptamer-attached Tds. Ap3-L-Td possessing well-organized multivalent aptamers provided higher uptake signals than Ap3-D-Td (Fig. 3c and d). The difference in the uptake efficiency between the L- and the D-based constructs was greater in the cancer cell line. This is likely due to the multivalent interaction between aptamers on L-Td and nucleolin, the target protein expressed on the surface of malignant cells.21 The Td construct also apparently contributed to the cellular uptake since the free aptamer was less delivered into cells than the single aptamer-labelled Tds (Ap1-L-Td, Fig. 3d). The analysis by flow cytometry exhibited that the delivery efficiency was improved in proportion to the number of aptamers on L-Td. In contrast, the efficiency was not considerably affected by the number of aptamers on D-Td.
Finally, the anti-proliferative effect by the multivalent aptamers on L-Td was examined by using a cell viability assay. Although all the aptamer-loaded Tds displayed dose-dependent toxicity in the cancer cell line at a certain level, the potency varied depending on the number of aptamers on Td and the backbone type of the DNA nanocarriers. Potency of Ap-L-Tds was higher than that of Ap-D-Tds in general and increased with the number of aptamers attached. Ap3-L-Td showed the highest potency leading to about 50% of HeLa cells remaining viable after treatment only with 500 nM of the trivalent aptamer on L-Td (red diamonds in Fig. 4a). However, more than 95% of the cells survived at the same concentration of the free AS1411 aptamer which has its EC50 value approximately at 30 μM (ESI, Fig. S7†), demonstrating that higher potency of Ap3-L-Td was obtained from higher uptake of aptamers based on the multivalent effect. Well-organized display of multivalent aptamers was important in delivery of aptamers and the subsequent downstream efficacy in cells, since irregularly arranged aptamers in Ap-D-Tds were not as potent as those in Ap-L-Tds (blue diamonds vs. red diamonds in Fig. 4a). Besides of known stability of the AS1411 aptamer in serum,21 enhanced serum stability and intracellular stability of L-Td also possibly contributed to the high potency of Ap-L-Tds. The moderately improved potency of Ap3-D-Td compared to that of Ap1-D-Td and Ap2-D-Td might be attributed to the higher aptamer portion in the construct (blue diamonds vs. blue squares and triangles in Fig. 4a). When incubated with the normal cells, all Tds exhibited significantly decreased cytotoxicity. Interestingly, Ap3-D-Td was slightly more cytotoxic than Ap3-L-Td in the normal cells (blue diamonds vs. red diamonds in Fig. 4b). This might be due to unexpected off-target effects in cells raised by its misassembled structures. In comparison with the free aptamer, single aptamer-attached Tds provided higher cytotoxicity in the cancer cells, which is due to the uptake efficiency boosted by Td (squares vs. stars). Td itself with either D- or L-backbone, however, did not influence on cell viability, proving that the DNA nanoconstructs themselves are non-cytotoxic (circles in Fig. 4). Thus, the cytotoxicity of aptamer-loaded Tds resulted solely from the aptamer moiety.
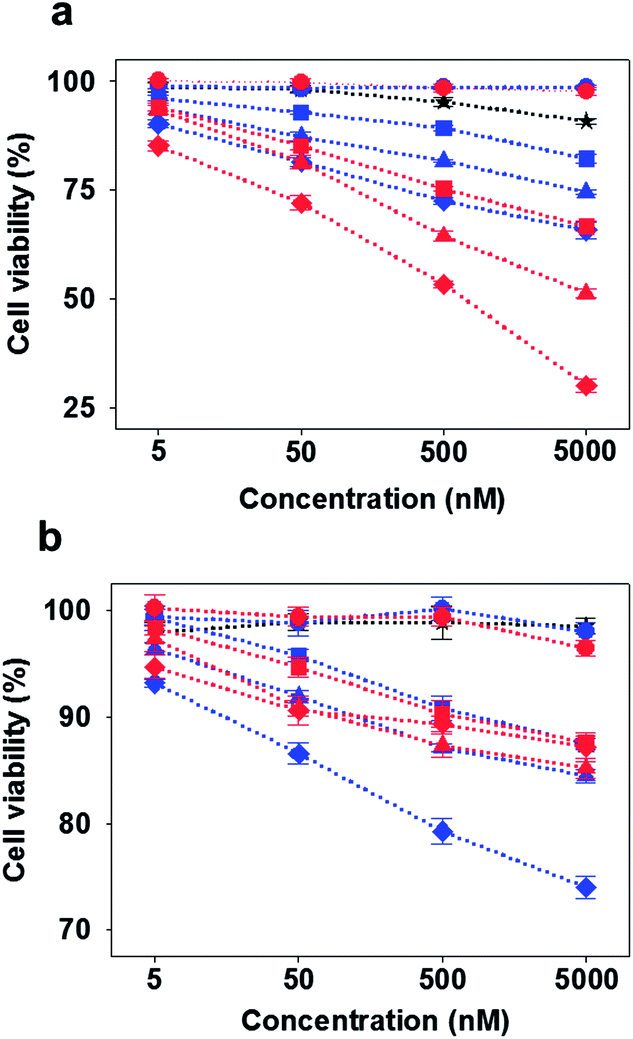 | ||
| Fig. 4 Cytotoxicity of D-DNA-based constructs (blue), L-DNA-based constructs (red), and free AS1411 aptamer (black) in (a) cancer and (b) normal cells. Circles: Td, squares: Ap1-Td, triangles: Ap2-Td, diamonds: Ap3-Td, star: free AS1411. | ||
To minimize the effect from the misassembled structures, we attempted purification of Ap3-D-Td by extracting the correctly assembled band in PAGE. As shown in the PAGE analysis of the extract portion (ESI, Fig. S8a†), we found that a small portion of undesired structures was again formed, although the portion of Ap3-D-Td increased compared to the state before purification. This might be due to dynamic equilibrium between Ap3-D-Td and other misassembled constructs under the conditions we employed. Purification of Ap3-D-Td led to improved intracellular delivery of the construct as shown by the analysis using a flow cytometer (ESI, Fig. S8b†); the uptake efficiency of the refined sample was higher than that of unpurified mixture. Compared with the mixture state, the refined portion showed higher cytotoxicity against the cancer cell line and similar cytotoxicity against the normal cell line, respectively (ESI, Fig. S8c†). When compared with L-Td constructs, the refined Ap3-Td still showed lower uptake in both normal and cancer cells (ESI, Fig. S8b†).
In conclusion, we have newly prepared a DNA tetrahedron based on L-sugar backbone. Compared with the natural D-sugar-based one, L-Td could be more easily delivered without using any transfection agent and showed higher nuclease resistance in serum. Utilizing the base-paring orthogonality between D-DNA and L-DNA, we could successfully assemble aptamer-loaded L-Tds whereas we failed to construct well-defined aptamer-attached D-Tds due to the cross-interaction between the bases in aptamer and those in D-Td. Compared with the free aptamer, the multivalent aptamers arranged on L-Td were found to show a 60-fold increase in potent cytotoxicity only against cancer cells, possibly because of enhanced uptake specifically into cancer cells, which display the target protein of the aptamer on the cell surface. In addition, proper arrangement of multivalent aptamers also contributed to their targeted potency, maximizing desired cytotoxicity in cancer cells and minimizing undesired cytotoxicity in normal cells. Although DNA tetrahedra have been proved to be useful drug carriers with excellent biocompatibility and low cytotoxicity in previous studies, we have demonstrated that nucleotide-based cargos have a potential to disturb the self-assembly of DNA nanoconstructs in this study. Therefore, our strategy employing L-DNA-based nanocarriers for delivery of therapeutic nucleic acids without altering thermodynamic properties of the assembly would be a promising solution to circumvent the problematic situation stemming from the disruption of desired base-pairing.
Acknowledgements
This study was supported by an intramural research program of KIST (Global RNAi Carrier Initiative), the National Research Foundation of Korea (NRF) grant funded by the Korean government (2010-0028684), a grant of the Basic Science Research Program (2011-0009172), a grant of the Proteogenomic Research Program through the National Research Foundation of Korea funded by the Ministry of Education, and a grant of the Korea Healthcare technology R&D Project, the Ministry of Health & Welfare, Republic of Korea (A121191).Notes and references
- K. R. Kim, D. R. Kim, T. Lee, J. Y. Yhee, B. S. Kim, I. C. Kwon and D. R. Ahn, Chem. Commun., 2013, 49, 2010 RSC.
- Q. Jiang, C. Song, J. Nangreave, X. Liu, L. Lin, D. Qiu, Z. G. Wang, G. Zou, X. Liang, H. Yan and B. Ding, J. Am. Chem. Soc., 2012, 134, 13396 CrossRef CAS PubMed.
- D. Smith, V. Schüller, C. Engst, J. Rädler and T. Liedlnomedicine, Nanomedicine, 2013, 8, 105 CrossRef CAS PubMed.
- S. Walsh, H. Yin, C. M. Erben, M. J. A. Wood and A. J. Turberfield, ACS Nano, 2011, 5, 5427 CrossRef PubMed.
- G. D. Hamblin, K. M. M. Carneiro, J. F. Fakhoury, K. E. Bujold and H. F. Sleiman, J. Am. Chem. Soc., 2012, 134, 2888 CrossRef CAS PubMed.
- M. Chang, C. S. Yang and D. M. Huang, ACS Nano, 2011, 5, 6156 CrossRef CAS PubMed.
- J. W. Keum, J. H. Ahn and H. Bermudez, Small, 2011, 7, 3529 CrossRef CAS PubMed.
- J. Li, H. Pei, B. Zhu, L. Liang and M. Wei, ACS Nano, 2011, 5, 8783 CrossRef CAS PubMed.
- K. Mohri, M. Nishikawa, N. Takahashi, T. Shiomi, N. Matsuoka, K. Ogawa, M. Endo, K. Hidaka, H. Sugiyama, Y. Takahashi and Y. Takakura, ACS Nano, 2012, 6, 5931 CrossRef CAS PubMed.
- X. Liu, Y. Xu, T. Yu, C. Clifford, Y. Liu, H. Yan and Y. Chang, Nano Lett., 2012, 12, 4254 CrossRef CAS PubMed.
- H. Lee, A. K. R. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman, C. G. Peng, K. Charisse, A. Borodovsky, M. Manoharan, J. S. Donahoe, J. Truelove, M. Nahrendorf, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2012, 7, 389 CrossRef CAS PubMed.
- J. B. Lee, J. Hong, D. K. Bonner, Z. Poon and P. T. Hammond, Nat. Mater., 2012, 11, 316 CrossRef CAS PubMed.
- J. Hunziker, H. J. Roth, M. Böhringer, A. Giger, U. Diederichsen, M. Göbel, R. Krishnan, B. Jaun, C. Leumann and A. Eschenmoser, Helv. Chim. Acta, 1993, 76, 259 CrossRef CAS.
- Z. Yang, A. M. Sismour, P. Sheng, N. L. Puskar and S. A. Benner, Nucleic Acids Res., 2007, 35, 4238 CrossRef CAS PubMed.
- C. Switzer, S. E. Moroney and S. A. Benner, J. Am. Chem. Soc., 1989, 111, 8322 CrossRef CAS.
- S. Pitsch, R. Krishnamurthy, M. Bolli, S. Wendeborn, A. Holzner, M. Minton, C. Lesueur, I. Schlonvogt, B. Jaun and A. Eschenmoser, Helv. Chim. Acta, 1995, 78, 1621 CrossRef CAS.
- S. Fujimori, K. Shudo and Y. Hashimoto, J. Am. Chem. Soc., 1990, 112, 7436 CrossRef CAS.
- D. J. Anderson, R. J. Reischer, A. J. Taylor and W. J. Wechter, Nucleosides Nucleotides, 1984, 3, 499 CAS.
- P. Tran, R. Moriyama, A. Maruyama, B. Rayner and J.-L. Mergny, Chem. Commun., 2011, 47, 5437 RSC.
- R. P. Goodman, R. M. Berry and A. J. Turberfield, Chem. Commun., 2004, 1372 RSC.
- P. J. Bates, D. A. Laber, D. M. Miller, S. D. Thomas and J. O. Trent, Exp. Mol. Pathol., 2009, 86, 151 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional data. See DOI: 10.1039/c3sc52601a |
| This journal is © The Royal Society of Chemistry 2014 |