A pillar[5]arene/imidazolium [2]rotaxane: solvent- and thermo-driven molecular motions and supramolecular gel formation†
Shengyi
Dong
a,
Jiayin
Yuan
b and
Feihe
Huang
*a
aState Key Laboratory of Chemical Engineering, Department of Chemistry, Zhejiang University, 310027 Hangzhou, P. R. China. E-mail: fhuang@zju.edu.cn; Fax: +86-571-8795-3189; Tel: +86-571-8795-3189
bMax Planck Institute of Colloids and Interfaces, Potsdam 14476, Germany
First published on 20th September 2013
Abstract
Based on the pillar[5]arene/imidazolium recognition motif, a [2]rotaxane was effectively prepared. Solvent/temperature triggered molecular motions of the pillar[5]arene ring on the imidazolium axle were successfully realized. By comparison of proton NMR spectra of the [2]rotaxane in different solvents, we found that if we increased the solvent polarity, the pillar[5]arene ring gradually moved away from the imidazolium part. In DMSO, we also could adjust the binding site of the pillar[5]arene ring by changing the temperature. Furthermore, in DMSO, the [2]rotaxane self-assembled to form a supramolecular gel, which showed multiple stimuli-responsiveness.
Introduction
Molecular motions are quite common in biosystems and closely related to macroscopic motions.1 Inspired by the dynamic and reversible nature of natural supramolecular assemblies, scientists have applied artificial supramolecular architectures to mimic and control molecular motions.2 Rotaxanes, considered as a typical type of mechanically interlocked structures, are crucial precursors for the fabrication of advanced supramolecular architectures3 and have been widely used as building blocks to fabricate molecular shuttles and switches which show molecular motions induced by certain stimuli.4 Though many well-known molecular recognition motifs,5 such as crown ether/ammonium salt recognition motifs, have been applied to investigate rotaxane-based molecular motions, the introduction of new molecular recognition motifs to the realm of rotaxanes will no doubt expand the applications of rotaxanes and show some unique stimuli-responsiveness.6Pillar[5]arenes,7 a new kind of macrocyclic host, were first reported in 2008 and have achieved quick development over the past five years.8 Though pillar[5]arenes have become among the most promising candidates to prepare supramolecular assemblies, such as supramolecular polymers,8b micelles8f and nano fibers,8e their application in molecular motions still remains limited.6c,7g,8h In our previous work, we found that a pillar[5]arene can complex an imidazolium salt with a high binding constant and form a [2]pseudorotaxane.9 Considering the rigid and pillar-like structures and host–guest properties of pillar[5]arenes, we wonder whether it is feasible to utilize this pillar[5]arene/imidazolium recognition motif to investigate pillar[5]arene-based reversible molecular motions. Here we designed and synthesized a pillar[5]arene-based [2]rotaxane and studied the temperature/solvent-controllable molecular motions of the pillar[5]arene ring on the imidazolium axle.
Results and discussion
As shown in Scheme 1, DEP5 (1,4-diethoxypillar[5]arene) and the semi-blocked rod-like component 1 were mixed together for two hours at low temperature (in an ice-salt bath), then 1-isocyanato-3,5-dimethylbenzene (2), which acted as the stopper, and dibutyltindilaurate (DBTDL) were added.10 This reaction gave [2]rotaxane 4 in quite a high yield, 92%. No dumbbell-shaped component 3 was observed in this reaction as monitored by TLC.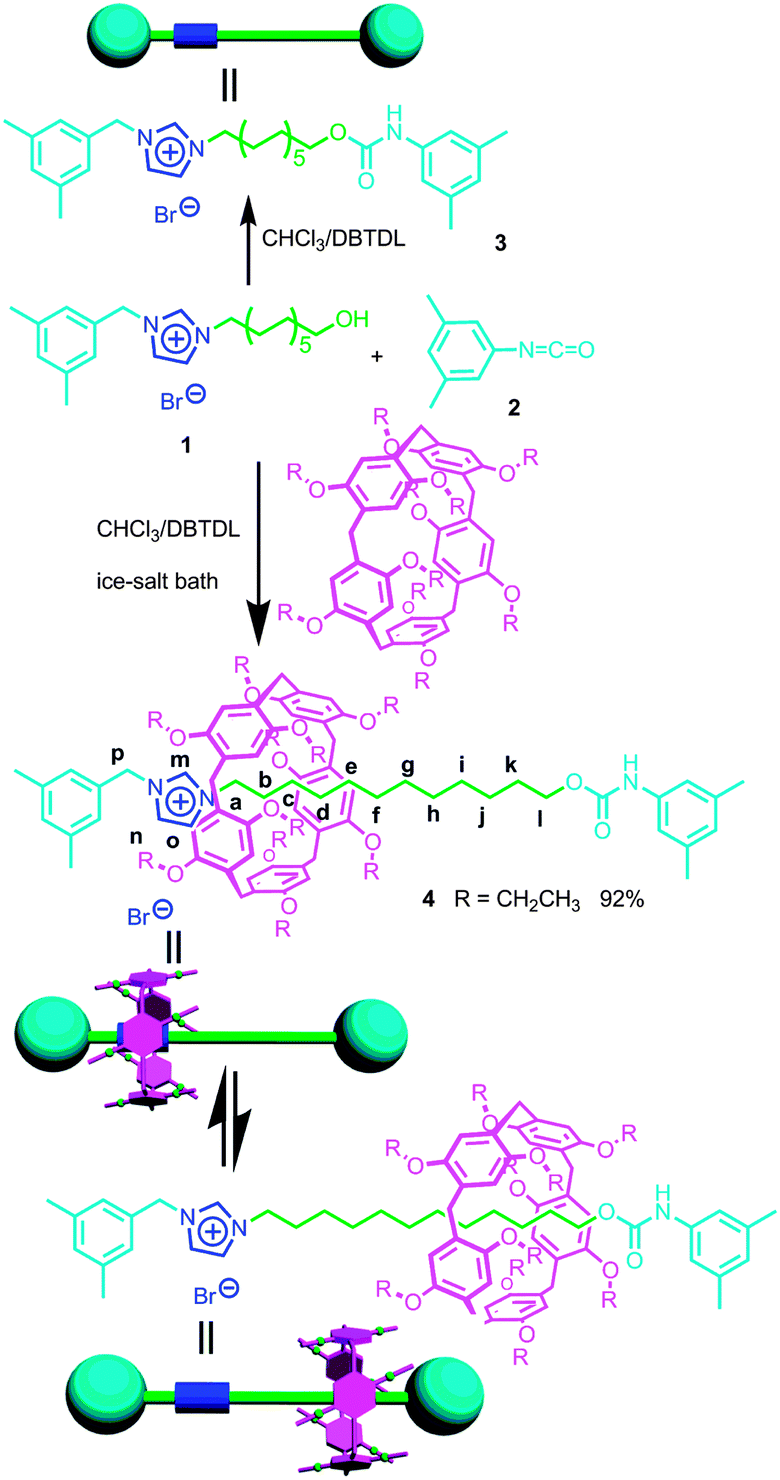 | ||
| Scheme 1 Synthetic route to [2]rotaxane 4 and the corresponding dumbbell-shaped component 3 and their cartoon representations. | ||
Considering the high binding capacity between pillar[5]arenes and imidazolium salts,9 we estimated that the imidazolium part and the adjacent methylene groups would be located in the cavity of the pillar[5]arene ring. Indeed, the 1H NMR spectrum and 2D NOESY spectrum of [2]rotaxane 4 in chloroform-d confirmed our deduction (Fig. 1, S7 and S12†). In chloroform-d, we found that due to the shielding effect, the signals of protons on the imidazolium part and adjacent methylene protons Hm, Hb, Hc and Hd shifted upfield obviously (Fig. 1). For example, the signals of protons Hm shifted from 10.77 ppm to 8.11 ppm and the signal of protons Hb and Hc showed chemical shifts below even 0 ppm. These changes were quite in accordance with previously reported pillar[5]arene-based threaded structures with alkyl chains,7h,8b,g,9 which meant that the imidazolium part and adjacent methylene groups were covered by the aromatic cavity of the pillar[5]arene ring. The NOESY spectrum of 4 in chloroform showed cross-signals between the aromatic protons of the pillar[5]arene ring and the benzyl protons Hp, indicating the formation of the host–guest inclusion complex (Fig. S12†).
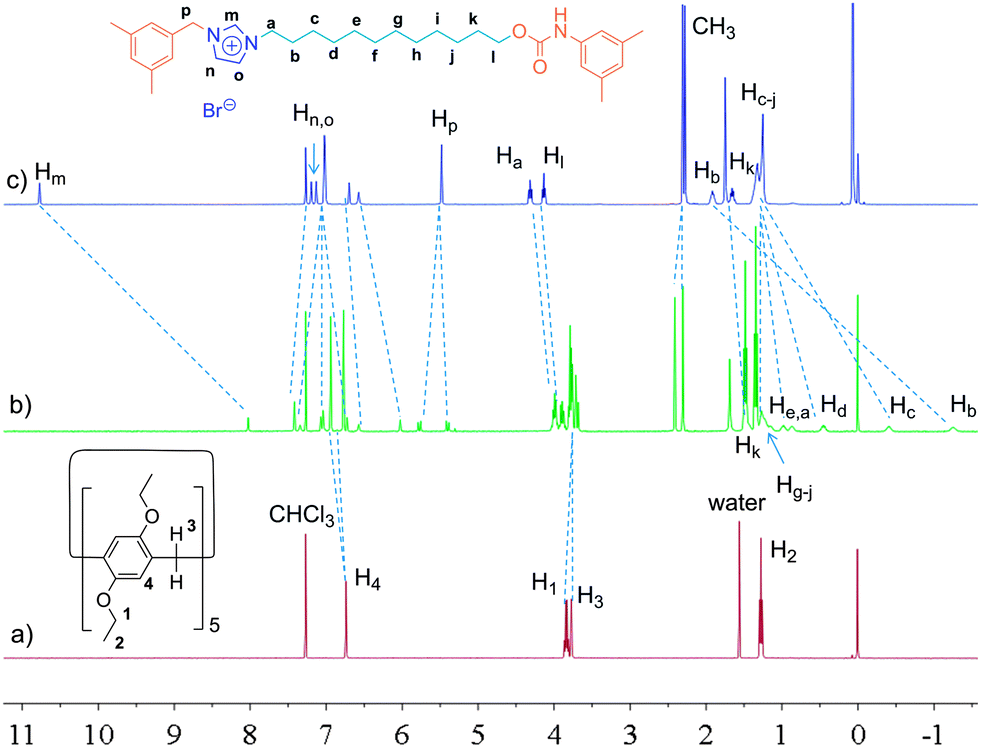 | ||
| Fig. 1 1H NMR spectra (400 MHz, chloroform-d, 25 °C) of 2.00 mM DEP5 (a), 2.00 mM [2]rotaxane 4 (b), and 2.00 mM dumbbell-shaped component 3 (c). | ||
In order to confirm this mechanically interlocked structure, a polar solvent, DMSO-d6, was used for proton NMR investigations. In DMSO-d6, we observed that due to the shielding effect, the signals of protons on the alkyl chains displayed upfield chemical shifts, which indicated the formation of a mechanically interlocked structure (Fig. 2). From 1H NMR spectra in DMSO-d6, we found that the signals of protons Hg, Hh, Hi and Hj moved upfield and all had chemical shifts below 0 ppm, which meant these protons were located in the cavity of the pillar[5]arene ring. For example, Hg,j and Hh,i shifted from 1.10 ppm to −0.12 and −0.41 ppm, respectively. The NOESY spectrum of 4 in DMSO-d6 showed obvious cross-signals between methylene protons H1 and H3 and phenyl protons H4 of the pillar[5]arene ring and the signal of methylene protons Hk of the alkyl chain (Fig. S13†). Compared with the 1H NMR spectrum of [2]rotaxane 4 in chloroform-d (Fig. 1), we found that in DMSO-d6, the pillar[5]arene ring was on average located on the methylene groups close to the carbamic stopper instead of the imidazolium unit. A possible reason for this was that DMSO-d6 could destroy the interactions between the pillar[5]arene ring and the imidazolium moiety and showed a strong solvophobic effect for the alkyl chain close to the stopper. From the difference between the binding sites, we thought that the addition of polar solvent molecules would not only destroy the hydrogen bonds between the pillar[5]arene ring and the imidazolium guest unit but also drag the imidazolium unit out of the pillar[5]arene cavity because the imidazolium guest unit is solvophilic when its counterion is the bromide anion. This result is similar to the findings of Gibson et al. with crown ether–polyurethane systems11a–e and cyclobis(p-xylyleneparaquat)–polyurethanes.11f Hence we thought that we could realize molecular motions through changing the solvent polarity.2a,d,6c,8h,9
 | ||
| Fig. 2 1H NMR spectra (400 MHz, DMSO-d6, 25 °C) of 2.00 mM DEP5 (a), 2.00 mM [2]rotaxane 4 (b), and 2.00 mM dumbbell-shaped component 3 (c). | ||
We changed the volume ratio of chloroform-d/DMSO-d6 to adjust the solvent polarity and control the molecular motion of the pillararene ring on the imidazolium axle. In Fig. 3, we found that as the solvent polarity increased due to the stepwise addition of DMSO-d6 to chloroform-d, the signals of the protons Hb, Hc and Hd, which were close to the imidazolium unit, were all moved downfield. On the contrary, the signals of the protons near to the carbamic stopper, such as Hg, Hh, Hi and Hj, shifted upfield. For example, the signal of proton Hb moved downfield from −1.27 ppm (chloroform-d) to −0.58 ppm (chloroform-d/DMSO-d6 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), −0.14 ppm (chloroform-d/DMSO-d6 = 1:5), 0.09 ppm (chloroform-d/DMSO-d6 = 1:7), 0.19 ppm (chloroform-d/DMSO-d6 = 1:9) and 0.38 ppm (DMSO-d6). On the other hand, the signal of proton Hi moved upfield from 1.27 ppm (choroform-d) to 1.06 ppm (chloroform-d/DMSO-d6 = 1:2), 0.49 ppm (chloroform-d/DMSO-d6 = 1:3), 0.24 ppm (chloroform-d/DMSO-d6 = 1:5), −0.26 ppm (chloroform-d/DMSO-d6 = 1:9) and −0.53 ppm (DMSO-d6). Similar chemical shift changes were also observed for protons Ha and Hk (Fig. S14†). From these observations, it was demonstrated that the imidazolium unit and the adjacent methylene groups gradually moved out of the cavity of the pillar[5]arene ring; meanwhile the pillar[5]arene ring moved onto the methylene groups close to the carbamic stopper. Upon changing the solvent polarity, the pillar[5]arene ring was located at different places on the axle on average. These observations indicated that as the solvent polarity changed, [2]rotaxane 4 showed the corresponding molecular motions and acted as a molecular shuttle.2a,d,6c,8h
:3), −0.14 ppm (chloroform-d/DMSO-d6 = 1:5), 0.09 ppm (chloroform-d/DMSO-d6 = 1:7), 0.19 ppm (chloroform-d/DMSO-d6 = 1:9) and 0.38 ppm (DMSO-d6). On the other hand, the signal of proton Hi moved upfield from 1.27 ppm (choroform-d) to 1.06 ppm (chloroform-d/DMSO-d6 = 1:2), 0.49 ppm (chloroform-d/DMSO-d6 = 1:3), 0.24 ppm (chloroform-d/DMSO-d6 = 1:5), −0.26 ppm (chloroform-d/DMSO-d6 = 1:9) and −0.53 ppm (DMSO-d6). Similar chemical shift changes were also observed for protons Ha and Hk (Fig. S14†). From these observations, it was demonstrated that the imidazolium unit and the adjacent methylene groups gradually moved out of the cavity of the pillar[5]arene ring; meanwhile the pillar[5]arene ring moved onto the methylene groups close to the carbamic stopper. Upon changing the solvent polarity, the pillar[5]arene ring was located at different places on the axle on average. These observations indicated that as the solvent polarity changed, [2]rotaxane 4 showed the corresponding molecular motions and acted as a molecular shuttle.2a,d,6c,8h
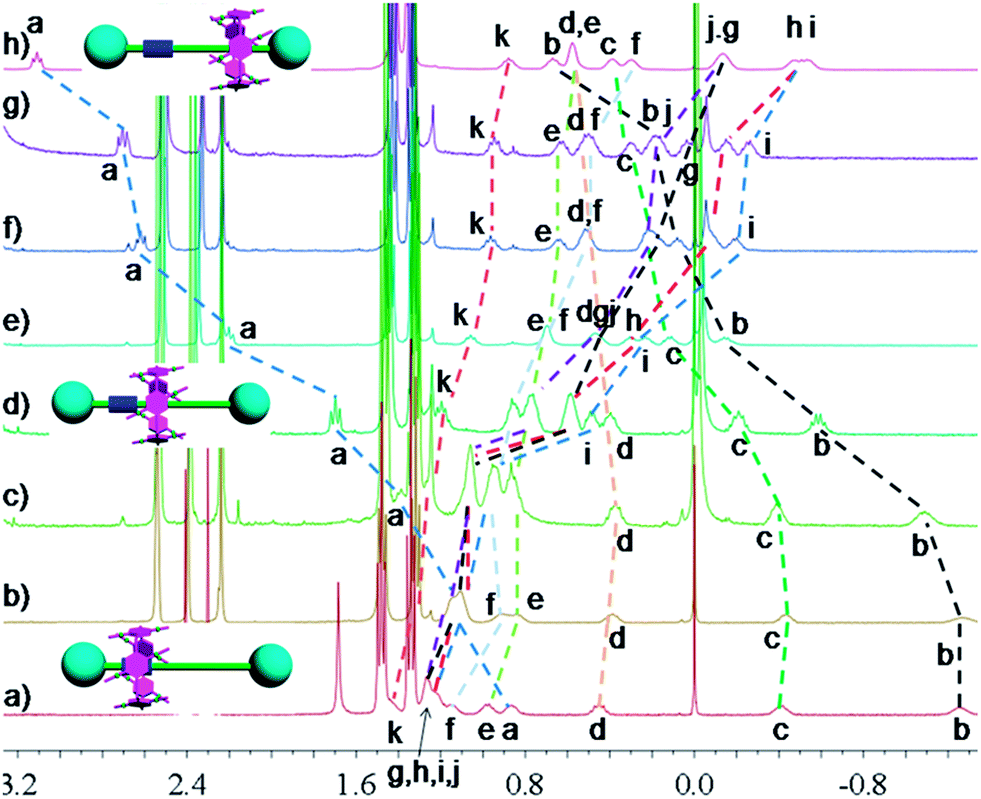 | ||
| Fig. 3
1H NMR spectra (400 MHz, 25 °C) of [2]rotaxane 4 in solvents with different polarities: (a) chloroform-d; (b) 1:1 chloroform-d/DMSO-d6; (c) 1:2 chloroform-d/DMSO-d6; (d) 1:3 chloroform-d/DMSO-d6; (e) 1:5 chloroform-d/DMSO-d6; (f) 1:7 chloroform-d/DMSO-d6; (g) 1:9 chloroform-d/DMSO-d6; (h) DMSO-d6. | ||
In nature and biological systems, relative molecular motions are very common and mostly induced by different stimuli, such as temperature, concentrations or electrical signals.5 Inspired by the molecular motions induced by the solvent polarity, here we further realized the rotaxane-based molecular motions in DMSO-d6 by changing the temperature. By applying temperature-dependent 1H NMR, we observed these molecular motions (Fig. 4 and 5). As the temperature increased, the signals of the methylene protons Hg, Hh, Hi, Hj, and Hk, which were close to the carbamic stopper, gradually moved downfield. For example, due to the deshielding effect, the chemical shift of proton Hh shifted from −0.44 ppm at 25 °C to −0.20 ppm at 35 °C, 0.35 ppm at 65 °C, and 0.82 ppm at 105 °C. In contrast, the signals of methylene protons Ha, Hb, Hc and Hd, which are close to the imidazolium unit, all showed upfield movements and the signals of protons Hc and Hd appeared at chemical shifts below 0 ppm at about 65 °C. With the further increase of temperature, these signals (Hb, Hc and Hd) shifted more and more upfield. For example, the signal of proton Hc shifted from −0.17 at 65 °C to −0.63 at 105 °C. In Fig. 4, we also observed that as the temperature increased, the signal of proton Ha moved upfield, along with the gradual downfield movement of the signal of proton Hk (Fig. 4 and 5). From these 1H NMR results, it was demonstrated that at low temperature, due to the high polarity of DMSO-d6, the pillar[5]arene ring was on the methylene groups close to the carbamic stopper, which caused a shielding effect of the alkyl chain near the carbamic stopper. With the increase of temperature, the pillararene cavity gradually slid to the imidazolium unit and made the signals of the protons on the imidazolium unit and adjacent methylene groups moved upfield. By comparing the 1H NMR spectra of [2]rotaxane 4 at 25 °C and 115 °C, it was indicated that heating and cooling effectively controlled the binding sites and drove the shuttle-like molecular motions. A possible reason for these observations was that the relative stabilities of the pillar[5]arene/imidazolium complexation and the pillar[5]arene/alkyl chain binding were quite different and had a temperature dependence, which meant that at different temperatures the pillar[5]arene ring favored moving to different binding sites to form a more stable complex.2c From these results, we think that [2]rotaxane 4 was proven to be a good candidate for stimuli-controlled molecular shuttles and would have great potential application in investigating molecular motions.2b,c,6c,8g,10c
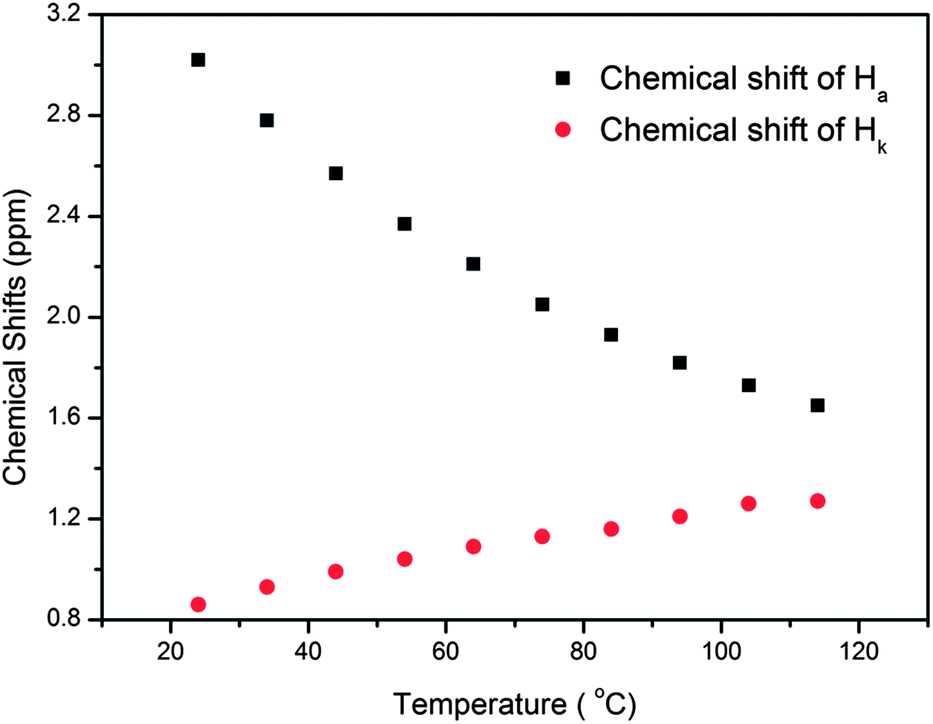 | ||
| Fig. 4 Temperature-dependent chemical shifts of Ha and Hk (400 MHz, DMSO-d6). | ||
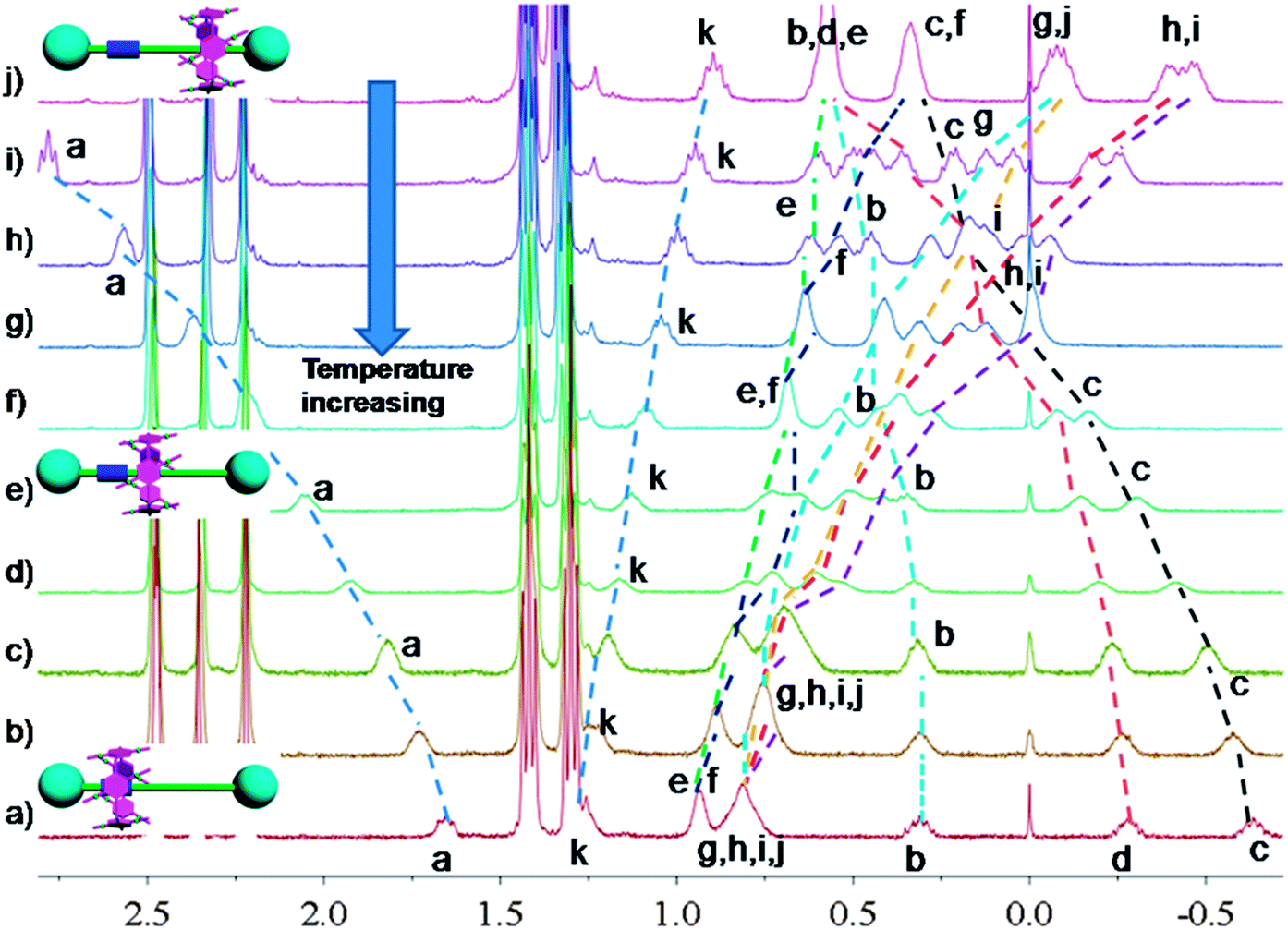 | ||
| Fig. 5 Partial variable-temperature 1H NMR of [2]rotaxane 4 (400 MHz, DMSO-d6): (a) 115 °C; (b) 105 °C; (c) 95 °C; (d) 85 °C; (e) 75 °C; (f) 65 °C; (g) 55 °C; (h) 45 °C; (i) 35 °C; (j) 25 °C. | ||
Surprisingly, we found that [2]rotaxane 4 formed supramolecular gels in DMSO or DMSO/water (5:1), while the dumbbell-shaped component 3 could not form supramolecular gels under the same conditions. Interestingly, under normal conditions these supramolecular gels were quite stable for at least half a year. Xerogels prepared by freeze-drying a gel of 4 in DMSO were examined by scanning electron microscopy (SEM). SEM pictures revealed that [2]rotaxane 4 aggregated into fibers with diameters of 200 nm and lengths of several hundred μm (Fig. 6b–e). These SEM images showed that the amphiphilic gelator 4 (the imidazolium unit was solvophilic when its counterion was the bromide anion while the alkyl chain was solvophobic in DMSO) self-assembled at the nanoscale via intermolecular interactions to produce fibrils, and these fibrils further physically cross-linked and interwove together to form a dense net-like structure. By entrapping solvent molecules, supramolecular gels formed.12
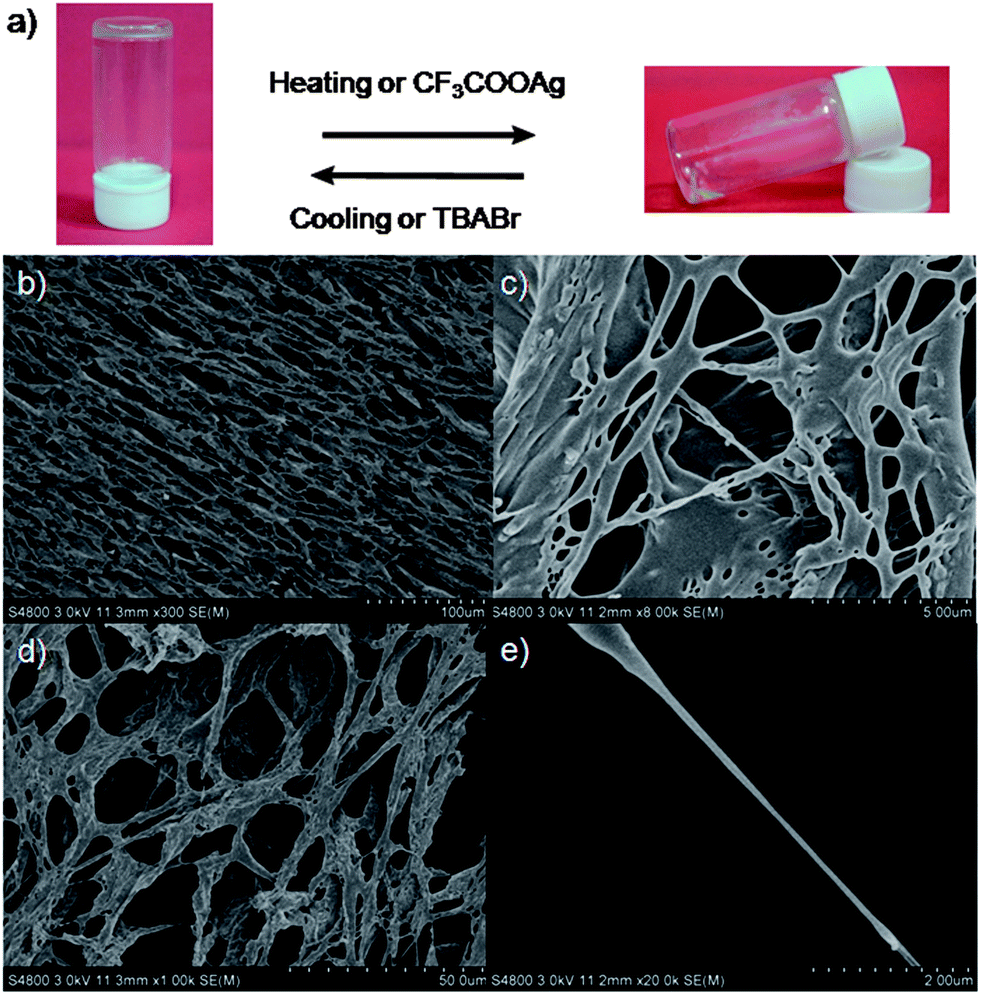 | ||
| Fig. 6 Stimuli-responsive reversible sol–gel transitions ((a), AgBr was removed) and SEM images of freeze-dried supramolecular gels (b–e). | ||
Considering the fact that reversibility and responsiveness are important features of supramolecular gels,12 we investigated stimuli-triggered reversible gel–sol transitions of the supramolecular gel 4/DMSO (Fig. 6a). After the addition of CF3COOAg (silver trifluoroacetate) to the supramolecular gel (Fig. 6a), we found that the gel gradually collapsed and finally became a transparent solution (AgBr was removed), while upon adding a little excess TBABr (tetrabutylammonium bromide) to the solution, we observed the reformation of the supramolecular gel. A presumed reason was that after ion exchange (from the bromide anion to the CF3COO anion), gelator 4 was amphiphilic, which led to the disappearance of the driving forces of gelation. After the addition of TBABr, the counterion was changed back to the bromide anion, leading to the reformation of the supramolecular gel. We also achieved a reversible sol–gel transition upon sequential addition of CF3SO3Ag and TBABr (Fig. 6a).12
As expected, we found that this supramolecular gel was quite sensitive to temperature. The transparent gel gradually became solution phase when temperature increased and recovered at room temperature. A quick reversible gel–sol transition was also achieved by shaking or resting. We also observed that this supramolecular gel gradually collapsed when treated with ultrasonic waves and reformed after standing.11
Conclusions
In conclusion, we prepared a pillar[5]arene-based [2]rotaxane. This [2]rotaxane showed solvent- and thermo-driven molecular motions. By adjusting the solvent polarity or changing the temperature, the pillar[5]arene ring can be on average located at different parts of the dumbbell-shaped component. These changes were monitored by 1H NMR. Molecular motions and multiple binding sites in the [2]rotaxane will no doubt endow it with the ability to generate supramolecular architectures with dynamic functions and properties. This [2]rotaxane further self-assembled into supramolecular gels under appropriate conditions. The supramolecular gels showed reversible gel–sol phase transitions upon heating/cooling, shaking/resting, or the addition of different anions. The work presented here demonstrates that the pillar[5]arene/imidazolium complex can be used as a building block to construct multiple supramolecular aggregates, from mechanically interlocked structures to supramolecular gels under different conditions.Acknowledgements
This work was supported by the National Basic Research Program (2013CB834502), the National Natural Science Foundation of China (91027006 and 21125417), the Fundamental Research Funds for the Central Universities (2012QNA3013) and an Alexander von Humboldt Fellowship. We thank Prof. Markus Antonietti from the Max Planck Institute of Colloids and Interfaces for his kind assistance.Notes and references
- (a) P. A. Kroon, M. Kainosho and S. I. Chan, Nature, 1975, 256, 582–584 CrossRef CAS; (b) H. Noji, R. Yasuda, M. Yoshida and K. Kinoshita, Nature, 1997, 386, 299–302 CrossRef CAS PubMed; (c) J. Howard, Nature, 1997, 389, 561–567 CrossRef CAS PubMed; (d) R. Dominguez, Y. Freyzon, K. M. Trybus and C. Cohen, Cell, 1998, 94, 559–571 CrossRef CAS.
- (a) A. S. Lane, D. A. Leigh and A. Murphy, J. Am. Chem. Soc., 1997, 119, 11092–11093 CrossRef CAS; (b) G. Bottari, F. Dehez, D. A. Leigh, P. J. Nash, E. M. Pérez, J. K. Y. Wong and F. Zerbetto, Angew. Chem., Int. Ed., 2003, 42, 5886–5889 CrossRef CAS PubMed; (c) J. V. Hernández, E. R. Kay and D. A. Leigh, Science, 2004, 306, 1532–1537 CrossRef PubMed; (d) D. A. Leigh, M. Á. F. Morales, E. M. Pérez, J. K. Y. Wong, C. G. Saiz, A. M. Z. Slawin, A. J. Carmichael, D. M. Haddleton, A. M. Brouwer, W. J. Buma, G. W. H. Wurpel, S. León and F. Zerbetto, Angew. Chem., Int. Ed., 2005, 44, 3062–3067 CrossRef CAS PubMed; (e) B. Lewandowski, G. D. Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed; (f) K. Zhu, V. N. Vukotic, N. Noujeim and S. J. Loeb, Chem. Sci., 2012, 3, 3265–3271 RSC; (g) L. Liu, Y. Liu, P. Liu, J. Wu, Y. Guan, X. Hu, C. Lin, Y. Yang, X. Sun, J. Ma and L. Wang, Chem. Sci., 2013, 4, 1701–1706 RSC; (h) C. G. Collins, E. M. Peck, P. J. Kramer and B. D. Smith, Chem. Sci., 2013, 4, 2557–2563 RSC.
- (a) Y. Liu, H. Wang, P. Liang and H.-Y. Zhang, Angew. Chem., Int. Ed., 2004, 43, 2690–2694 CrossRef CAS; (b) D.-H. Qu, Q.-C. Wang and H. Tian, Angew. Chem., Int. Ed., 2005, 44, 5296–5299 CrossRef CAS PubMed; (c) T. He and C.-F. Chen, J. Org. Chem., 2008, 73, 7735–7742 CrossRef PubMed; (d) V. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines-Concepts and Prospectives for Nano World, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed; (e) L. M. Klivansky, G. Koshkakaryan, D. Cao and Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 4185–4189 CrossRef CAS PubMed; (f) A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS PubMed; (g) M. Zhang, K. Zhu and F. Huang, Chem. Commun., 2010, 46, 8131–8141 RSC; (h) S. Li, B. Zheng, J. Chen, S. Dong, Z. Ma, F. Huang and H. W. Gibson, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4067–4073 CrossRef CAS; (i) W. Zhu, W. Li, C. Wang, J. Cui, H. Yang, Y. Jiang and G. Li, Chem. Sci., 2013, 4, 3583–3590 RSC.
- (a) F. Huang and H. W. Gibson, Prog. Polym. Sci., 2005, 30, 982–1018 CrossRef CAS PubMed; (b) Z. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024–6046 CrossRef CAS PubMed; (c) L. Fang, M. A. Olson, D. Benítez, E. Tkatchouk, W. A. Godddard and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17–29 RSC; (d) Z. Niu, F. Huang and H. W. Gibson, J. Am. Chem. Soc., 2011, 133, 2836–2839 CrossRef CAS PubMed; (e) D.-H. Qu and H. Tian, Chem. Sci., 2011, 2, 1011–1015 RSC; (f) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed; (g) V. N. Vukotic and S. J. Loeb, Chem. Soc. Rev., 2012, 41, 5896–5906 RSC; (h) C. B. Caputo, K. Zhu, V. N. Vukotic, S. J. Loeb and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 960–963 CrossRef CAS PubMed.
- (a) J. D. Badjić, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef PubMed; (b) W. C. Yount, H. Juwarker and S. L. Craig, J. Am. Chem. Soc., 2003, 125, 15302–15303 CrossRef CAS PubMed; (c) W. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am. Chem. Soc., 2006, 128, 11663–11672 CrossRef CAS PubMed; (d) S.-Y. Hsueh, C.-T. Kuo, T.-W. Lu, C.-C. Lai, Y.-H. Liu, H.-F. Hsu, S.-M. Peng, C.-h. Chen and S.-H. Chiu, Angew. Chem., Int. Ed., 2010, 49, 9170–9173 CrossRef CAS PubMed; (e) Y. Kohsaka, K. Nakazono, Y. Koyama, S. Asai and T. Takata, Angew. Chem., Int. Ed., 2011, 50, 4872–4875 CrossRef CAS PubMed.
- (a) W. Jiang, H. D. F. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852–13853 CrossRef CAS PubMed; (b) M. Zhang, S. Li, S. Dong, J. Chen, B. Zheng and F. Huang, Macromolecules, 2011, 44, 9629–9634 CrossRef CAS; (c) Z. Zhang, C. Han, G. Yu and F. Huang, Chem. Sci., 2013, 3, 3026–3031 RSC.
- (a) T. Ogoshi, S. Kanai, S. Fujinami, T.-a. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed; (b) D. Cao, Y. Kou, J. Liang, Z. Chen, L. Wang and H. Meier, Angew. Chem., Int. Ed., 2009, 48, 9721–9723 CrossRef CAS PubMed; (c) C. Li, Q. Xu, J. Li, F. Yao and X. Jia, Org. Biomol. Chem., 2010, 8, 1568–1576 RSC; (d) Z. Zhang, B. Xia, C. Han, Y. Yu and F. Huang, Org. Lett., 2010, 12, 3285–3287 CrossRef CAS PubMed; (e) W. Si, L. Chen, X.-B. Hu, G. Tang, Z. Chen, J.-H. Hou and Z.-T. Li, Angew. Chem., Int. Ed., 2011, 50, 12564–12568 CrossRef CAS PubMed; (f) G. Yu, C. Han, Z. Zhang, J. Chen, X. Yan, B. Zheng, S. Liu and F. Huang, J. Am. Chem. Soc., 2012, 134, 8711–8717 CrossRef CAS PubMed; (g) M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed; (h) T. Ogoshi, H. Kayama, D. Yamafuji, T. Aoki and T.-a. Yamagishi, Chem. Sci., 2012, 3, 3221–3226 RSC; (i) X. Shu, S. Chen, J. Li, Z. Chen, L. Weng, X. Jia and C. Li, Chem. Commun., 2012, 48, 2967–2969 RSC; (j) T. Ogoshi, D. Yamafuji, T. Aoki, K. Kitajima, T.-a. Yamagishi, Y. Hayashi and S. Kawauchi, Chem.–Eur. J., 2012, 18, 7493–7500 CrossRef CAS PubMed; (k) C. Li, J. Ma, L. Zhao, Y. Zhang, Y. Yu, X. Shu, J. Li and X. Jia, Chem. Commun., 2013, 49, 1924–1926 RSC; (l) H. Li, D.-X. Chen, Y.-L. Sun, Y. Zheng, L.-L. Tan, P. S. Weiss and Y.-W. Yang, J. Am. Chem. Soc., 2013, 135, 1570–1576 CrossRef CAS PubMed; (m) H. Zhang, X. Ma, J. Guo, K. T. Nguyen, Q. Zhang, X.-J. Wang, H. Yan, L. Zhu and Y. Zhao, RSC Adv., 2013, 3, 368–371 RSC; (n) S. Sun, X.-Y. Hu, D. Chen, J. Shi, Y. Dong, C. Lin, Y. Pan and L. Wang, Polym. Chem., 2013, 4, 2224–2229 RSC.
- (a) C. Li, L. Zhao, J. Li, X. Ding, S. Chen, Q. Zhang, Y. Yu and X. Jia, Chem. Commun., 2010, 46, 9016–9018 RSC; (b) Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397–1401 CrossRef CAS PubMed; (c) N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668–5671 CrossRef CAS PubMed; (d) X.-B. Hu, Z. Chen, G. Tang, J.-L. Hou and Z.-T. Li, J. Am. Chem. Soc., 2012, 134, 8384–8387 CrossRef CAS PubMed; (e) Y. Yao, M. Xue, J. Chen, M. Zhang and F. Huang, J. Am. Chem. Soc., 2012, 134, 15712–15715 CrossRef CAS PubMed; (f) G. Yu, M. Xue, Z. Zhang, J. Li, C. Han and F. Huang, J. Am. Chem. Soc., 2012, 134, 13248–13251 CrossRef CAS PubMed; (g) X.-Y. Hu, X. Wu, Q. Duan, T. Xiao, C. Lin and L. Wang, Org. Lett., 2012, 14, 4826–4829 CrossRef CAS PubMed; (h) Y. Guan, M. Ni, X. Hu, T. Xiao, S. Xiong, C. Lin and L. Wang, Chem. Commun., 2012, 48, 8529–8531 RSC; (i) L. Gao, B. Zheng, Y. Yao and F. Huang, Soft Matter, 2013, 9, 7314–7319 RSC.
- S. Dong, B. Zheng, Y. Yao, C. Han, J. Yuan, M. Antonietti and F. Huang, Adv. Mater., 2013, 25 DOI:10.1002/adma.201303652.
- (a) F. Wang, Q. Zhou, K. Zhu, S. Li, C. Wang, M. Liu, N. Li, F. R. Fronczek and F. Huang, Tetrahedron, 2009, 65, 1488–1494 CrossRef CAS PubMed; (b) S. Dong, C. Han, B. Zheng, M. Zhang and F. Huang, Tetrahedron Lett., 2012, 53, 3668–3671 CrossRef PubMed; (c) Z. Qi, P. M. Molina, W. Jiang, Q. Wang, K. Nowosinski, A. Schulz, M. Gradzielski and C. A. Schalley, Chem. Sci., 2012, 3, 2073–2082 RSC.
- (a) E. Marand, Q. Hu, H. W. Gibson and B. Veytsman, Macromolecules, 1996, 29, 2555–2562 CrossRef CAS; (b) C. Gong and H. W. Gibson, J. Am. Chem. Soc., 1997, 119, 8585–8591 CrossRef CAS; (c) C. Gong and H. W. Gibson, Angew. Chem., Int. Ed. Engl., 1997, 36, 2331–2333 CrossRef CAS; (d) C. Gong, T. E. Glass and H. W. Gibson, Macromolecules, 1998, 31, 308–313 CrossRef CAS; (e) C. Gong, Q. Ji, C. Subramaniam and H. W. Gibson, Macromolecules, 1998, 31, 1814–1818 CrossRef CAS; (f) P. E. Mason, W. S. Bryant and H. W. Gibson, Macromolecules, 1999, 32, 1559–1569 CrossRef CAS.
- (a) T. Fenske, H.-G. Korth, A. Mohr and C. Schmuck, Chem.–Eur. J., 2012, 18, 738–755 CrossRef CAS PubMed; (b) X. Yan, D. Xu, J. Chen, M. Zhang, B. Hu, Y. Yu and F. Huang, Polym. Chem., 2013, 4, 3312–3322 RSC; (c) D. Liu, D. Wang, M. Wang, Y. Zheng, K. Koynov, G. K. Auernhammer, H.-J. Butt and T. Ikeda, Macromolecules, 2013, 46, 4617–4625 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Compound characterization, full synthetic details and other materials. See DOI: 10.1039/c3sc52481g |
| This journal is © The Royal Society of Chemistry 2014 |