Conversion of high-spin iron(III)–alkylperoxo to iron(IV)–oxo species via O–O bond homolysis in nonheme iron models†
Seungwoo
Hong‡
ab,
Yong-Min
Lee‡
a,
Kyung-Bin
Cho
a,
Mi Sook
Seo
a,
Dayoung
Song
a,
Jihae
Yoon
a,
Ricardo
Garcia-Serres
c,
Martin
Clémancey
d,
Takashi
Ogura
e,
Woonsup
Shin
*b,
Jean-Marc
Latour
*f and
Wonwoo
Nam
*a
aDepartment of Chemistry and Nano Science, Department of Bioinspired Science, Center for Biomimetic System, Ewha Womans University, Seoul 120-750, Korea. E-mail: wwnam@ewha.ac.kr
bDepartment of Chemistry and Interdisciplinary Program of Integrated Biotechnology, Sogang University, Seoul 121-742, Korea. E-mail: shinws@sogang.ac.kr
cUniversity of Grenoble Alpes, LCBM, F-38054 Grenoble, France
dCNRS UMR 5249, LCBM, F-38054 Grenoble, France
ePicobiology Institute, Graduate School of Life Science, University of Hyogo, Hyogo 678-1297, Japan
fCEA, DSV, iRTSV, LCBM, PMB, F-38054 Grenoble, France. E-mail: jean-marc.latour@cea.fr
First published on 18th September 2013
Abstract
The mechanism of the alkylperoxo O–O bond cleavage of low-spin iron(III)–alkylperoxo species has been well established in nonheme iron models. In contrast, the alkylperoxo O–O bond cleavage in nonheme high-spin iron(III)–alkylperoxo species binding an axial ligand has yet to be elucidated. Herein, we report the synthesis and characterization of mononuclear nonheme high-spin iron(III)–alkylperoxo complexes each bearing an N-tetramethylated 13-membered macrocyclic ligand (13-TMC), [FeIII(OOC(CH3)3)(13-TMC)]2+ and [FeIII(OOC(CH3)2C6H5)(13-TMC)]2+. The high-spin iron(III)–alkylperoxo complexes were converted to an iron(IV)–oxo complex at a fast rate upon addition of thiocyanate (NCS−) via the formation of a short-lived intermediate. This intermediate was identified as a high-spin iron(III)–alkylperoxo complex binding a thiocyanate ion as an axial ligand by characterizing it with various spectroscopic methods and density functional theory (DFT) calculations. We have also provided strong evidence that conversion of the high-spin iron(III)–alkylperoxo complex to its corresponding iron(IV)–oxo complex occurs via O–O bond homolysis. Thus, we have concluded that the role of the axial ligand binding to a high-spin iron(III)–alkylperoxo complex is to facilitate the alkylperoxo O–O bond cleavage via the “push effect”, which has been well established in heme enzymes. To the best of our knowledge, the present study reports the first clear example showing the O–O bond homolysis of a high-spin iron(III)–alkylperoxo complex and the axial ligand effect on the alkylperoxo O–O bond cleavage in nonheme iron models.
Introduction
Iron–dioxygen adducts, including iron(III)–hydroperoxo (FeIII–OOH) and iron(III)–alkylperoxo (FeIII–OOCR3) species, have been identified as key intermediates in the catalytic cycles of dioxygen activation by heme and nonheme iron enzymes.1–3 The peroxide ligands of the iron(III)–hydroperoxo and –alkylperoxo species are cleaved either homolytically or heterolytically, resulting in the generation of high-valent iron(IV or V)–oxo intermediates as active oxidants that affect the oxidation of organic substrates. A notable example is the formation of the iron(IV)–oxo porphyrin π-cation radical species, referred to as Compound I (Cpd I), via heterolytic O–O bond cleavage of iron(III)–hydroperoxo intermediates in cytochromes P450 and their models (Scheme 1, eqn (1)).2 In the case of nonheme iron models, the formation of iron(IV)–oxo complexes via the O–O bond homolysis of iron(III)–hydroperoxo and –alkylperoxo species has been reported with spectroscopic evidence (Scheme 1, eqn (2)).4 Similarly, the homolytic O–O bond cleavage of nonheme manganese(III)–alkylperoxo complexes has been demonstrated recently.5 In addition to the peroxide O–O bond cleavage, iron(III)–hydroperoxo species undergo a protonation-assisted loss of H2O2via Fe–O bond cleavage (Scheme 1, eqn (3)). One example in nonheme iron enzymes is superoxide reductase (SOR), which reduces superoxide to hydrogen peroxide as part of the organism's natural defenses in anaerobic and microaerophilic organisms.6–8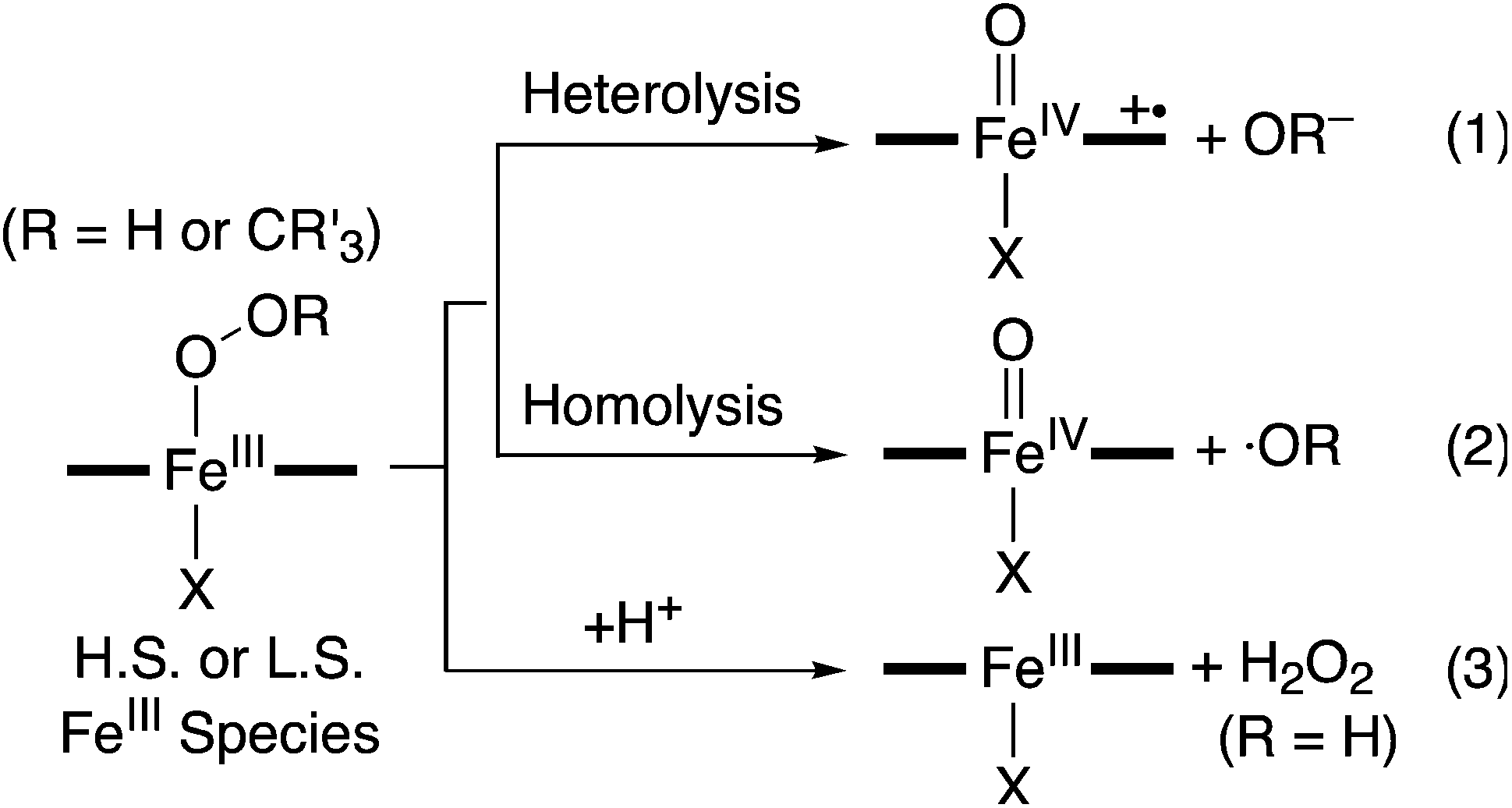 | ||
| Scheme 1 | ||
It has been well documented that the fate of the peroxide ligands of the Fe(III)–OOR(H) species is markedly affected by axial ligands in enzymatic and biomimetic reactions (e.g., heterolytic and homolytic O–O bond cleavage and H2O2 release as shown in Scheme 1).2,9 Spin states of the Fe(III)–OOR(H) species (e.g., high-spin (H.S.) and low-spin (L.S.) iron(III) ions) have also been considered as an important factor that modulates the strength of Fe–O and O–O bonds, thereby determining the reaction pathways such as the O–O bond vs Fe–O bond cleavage of nonheme Fe(III)–OOR(H) species (Scheme 1).10 For example, it has been shown that low-spin iron(III)–alkylperoxo complexes undergo homolytic O–O cleavage of the alkylperoxide ligands to form iron(IV)–oxo species (Scheme 1, eqn (2)),4b,10a whereas Fe–O bond cleavage occurs preferentially in high-spin iron(III)–alkylperoxo species (Scheme 1, eqn (3)).8b,c,9b However, the axial ligand effect on the O–O bond or Fe–O bond cleavage in high-spin iron(III)–alkylperoxo species has yet to be elucidated, although a number of high-spin iron(III)–alkylperoxo complexes have been synthesized and characterized well.8,11 Herein, we report the synthesis and characterization of mononuclear nonheme high-spin iron(III)–alkylperoxo complexes each bearing an N-tetramethylated 13-membered macrocyclic ligand (13-TMC = 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclotridecane).12 This high-spin iron(III)–alkylperoxo complex is converted to the corresponding iron(IV)–oxo complex upon binding of an anionic ligand. The iron(IV)–oxo product with the anionic axial ligand is fully characterized with various spectroscopic methods. The mechanism of the O–O bond cleavage of the high-spin iron(III)–alkylperoxo complex upon binding an anionic axial ligand is discussed as well.
Results and discussion
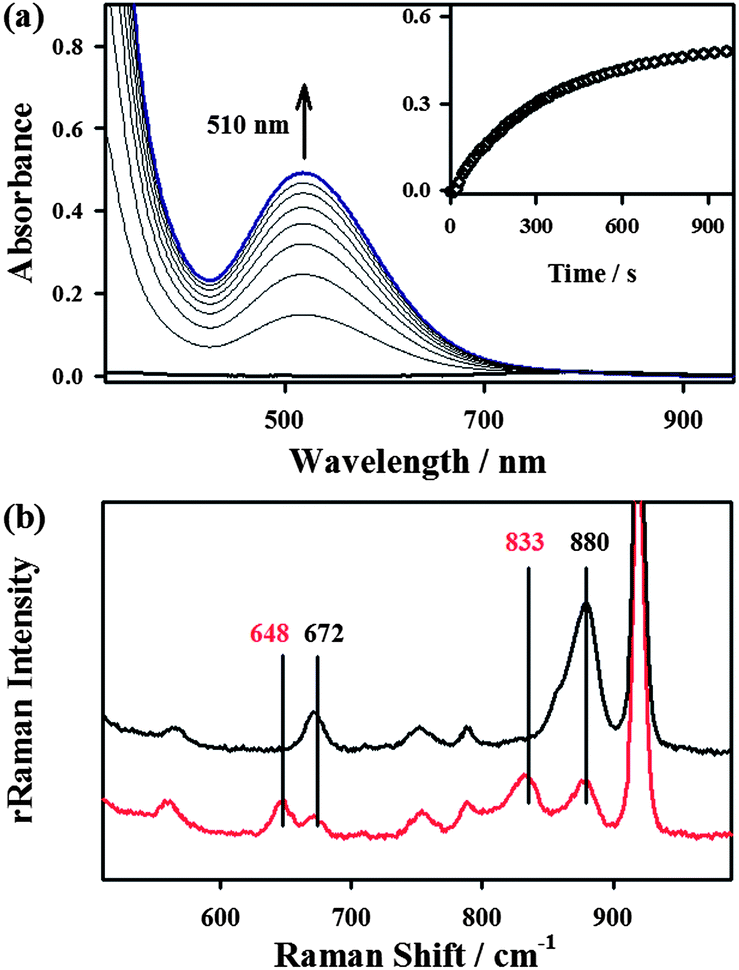
The starting iron(II) complex, FeII(13-TMC)(CF3SO3)2 (1), was synthesized by reacting equimolar amounts of FeII(CF3SO3)2 and 13-TMC in CH3CN under an inert atmosphere, as reported previously.13 Addition of alkyl hydroperoxides, such as tert-butyl hydroperoxide (t-BuOOH) and cumyl hydroperoxide (Cumyl-OOH), to 1 in CH3CN at −40 °C afforded the formation of purple intermediates, denoted as BH2 and CH2, respectively. The intermediates were metastable (t1/2 ∼ 1.5 h) under the reaction conditions and characterized with various spectroscopic methods, including UV-vis, electron paramagnetic resonance (EPR), Mössbauer, and resonance Raman (rRaman) spectroscopies.The UV-vis spectra of BH2 and CH2 exhibit maximum absorption bands at 510 nm (ε = 1100 M−1 cm−1) and 500 nm (ε = 1000 M−1 cm−1) (Fig. 1a for BH2 and ESI, Fig. S1† for CH2), respectively; these bands are assigned to an alkylperoxo-to-iron(III) charge transfer band (LMCT).8,11 The X-band EPR spectra of BH2 and CH2, recorded in a frozen CH3CN solution at 5 K, exhibit signals corresponding to high-spin (S = 5/2) iron(III) species with g values of 6.8, 5.4, and 2.0 for BH2 and 6.3, 5.5, and 2.1 for CH2 (ESI, Fig. S2†).8c,11b,c The Mössbauer spectrum of BH2, recorded at 4.2 K (Fig. 2a) in the absence of a strong applied magnetic field, displays a central doublet and a broad magnetic component, which can be interpreted as a single high-spin ferric species in two differing relaxation states (ESI, Fig. S3 and S4† for 1 and BH2, respectively). A small amount of unreacted 1 (8%, green line) is also present. The spectrum is difficult to simulate due to a distribution of internal fields. However, this distribution can be lifted by the application of a 7 T magnetic field that aligns the magnetic moments, giving rise to the six-line spectrum displayed in Fig. 2b, characteristic of a high-spin ferric ion. Most of the spectrum (∼90% of the absorption) could be simulated with parameters consistent with this assignment (ESI, Table S1†). It is noteworthy that this species possesses a large zero-field splitting in the range of those of high-spin ferric hemes and a small isomer shift more in line with a low-spin FeIII. However, attempts to simulate it as an S = 1/2 system were unsuccessful.
 | ||
| Fig. 1 (a) UV-vis spectrum of BH2 formed in the reaction of 1 (0.50 mM) and t-BuOOH (1.5 mM) in CH3CN at −40 °C. Inset shows the time trace monitored at 510 nm for the formation of BH2. (b) Resonance Raman spectra of [(13-TMC)FeIII(16O16OCumyl)]2+ (black line) and [(13-TMC)FeIII(18O18OCumyl)]2+ (red line) generated in the reactions of 1 (16 mM) and 3 equiv. of Cumyl-16O16OH and Cumyl-18O18OH (70% 18O enriched), respectively, in CH3CN at −40 °C (λex = 442 nm). | ||
 | ||
| Fig. 2 Mössbauer spectra of intermediates formed in the reactions of 1 and t-BuOOH before (a and b) and after (d and e) addition of NCS−, and an authentic compound 4 (c). Data was collected at 4.2 K in a parallel applied field of 0.06 T (a, c, and d) or 7 T (b and e). The colored lines represent theoretical simulations of individual species FeII (green), BH2 (pink), 4 (blue), and 3 (pink-dashed). The orange component is an impurity. The Mössbauer and zero-field splitting parameters used in the simulations are listed in ESI, Table S1.† They are derived from the applied-field studies represented in ESI, Fig. S4, S7, and S8.† | ||
The rRaman spectrum of CH2, obtained upon 442 nm excitation in CH3CN at −40 °C, shows two isotopically sensitive bands at 672 and 880 cm−1 (Fig. 1b). The peak at 672 cm−1 shifts to 648 cm−1 upon 18O-substitution with a 16,18Δ value of 24 cm−1 and is assigned to the Fe–O stretch.14 The peak at 880 cm−1 shifts to 833 cm−1 upon 18O-substitution with a 16,18Δ = 47 cm−1 and is the O–O stretch.14 One notable observation in the rRaman data is that the Fe–O stretch of CH2 is higher than those of the previously reported high-spin Fe(III)–OOR complexes (e.g., νFe–O of 612 cm−1 for [FeIII(OOC(CH3)3)(15-TMC)(OTf)]+; 15-TMC = 1,4,8,12-tetramethyl-1,4,8,12-tetraazacyclopentadecane, OTf− = CF3SO3−),8c,11 indicating that this high-spin iron(III)–alkylperoxo complex possesses a stronger Fe–O bond. Based on the spectroscopic characterization of the BH2 and CH2 intermediates, we conclude that high-spin iron(III)–alkylperoxo complexes, assigned as [FeIII(OOC(CH3)3)(13-TMC)]2+ for BH2 and [FeIII(OOC(CH3)2C6H5)(13-TMC)]2+ for CH2, were produced in the reactions of 1 and alkyl hydroperoxides (Scheme 2, reaction a).
 | ||
| Scheme 2 | ||
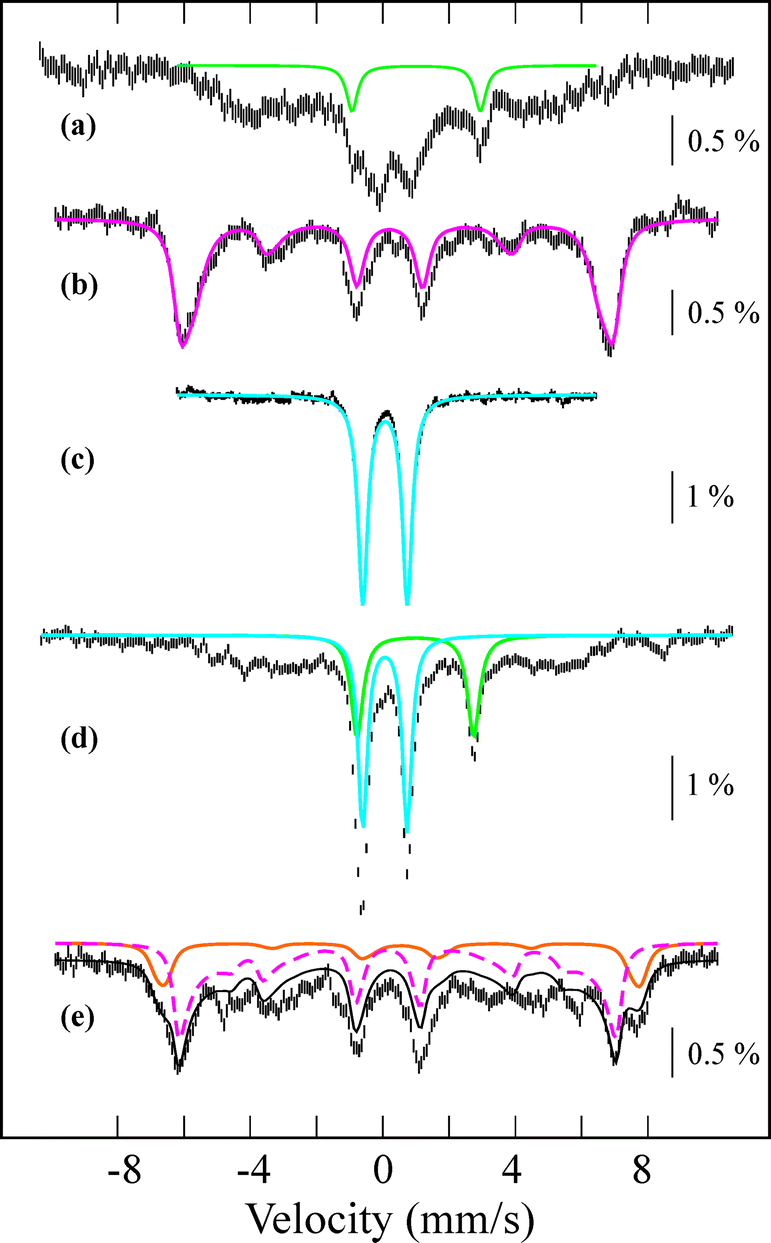
Interestingly, the addition of 1.2 equiv. of thiocyanate (NCS−) to the solutions of BH2 and CH2 yielded a green intermediate 4 with a broad electronic absorption band at 755 nm (Fig. 3 and ESI, S5†). The full conversion of BH2 to 4 was found to require one equiv. of thiocyanate by carrying out a titration experiment (Fig. 3, inset). The intermediate 4 with a characteristic absorption band of nonheme iron(IV)–oxo complexes15,16 was assigned unequivocally as [FeIV(O)(13-TMC)(NCS)]+ (Scheme 2, structure 4) by characterizing it with various spectroscopic methods and comparing the spectroscopic data with those of an authentic iron(IV)–oxo compound prepared independently (vide infra).
 | ||
| Fig. 3 UV-vis spectral changes showing the conversion of BH2 (blue, 0.50 mM) to 4 (green) with respect to the amount of thiocyanate added (0.20, 0.40, 0.60, 0.80, 1.0, and 1.2 equiv.) in CH3CN at −40 °C. Inset shows the absorbance recorded at 510 nm (blue circle) and 755 nm (green circle). Two horizontal dashed pink lines and one vertical dashed red line indicate the two saturation values and 1.0 equiv. of NCS− added, respectively. | ||
The authentic compound, [FeIV(O)(13-TMC)(NCS)]+ (4), was synthesized as follows: the reaction of 1 with 3 equiv. of iodosylbenzene (PhIO) in CH3CN at −40 °C afforded a green intermediate which was identified as [FeIV(O)(13-TMC)]2+ (6), as reported previously (Scheme 2, reaction f) (See ESI, Fig. S6† for the electrospray ionization mass spectrometry (ESI MS), UV-vis, and rRaman spectroscopy).13 The authentic compound 4 was then prepared by adding 1.2 equiv. of NCS− to the solution of 6 in CH3CN at −40 °C (Scheme 2, reaction g), by following literature methods for the synthesis of axial ligand-substituted [FeIV(O)(14-TMC)(X)]+ complexes (14-TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane; X− = NCS− and N3−).15,17 The authentic compound 4 was characterized with UV-vis, ESI MS, rRaman, and Mössbauer, and the spectroscopic data of the authentic compound 4 were compared with those of the product 4 formed in the reaction of BH2 and NCS−. The UV-vis spectrum of 4 exhibits an absorption band at λmax = 755 nm (ε = 160 M−1 cm−1) (ESI, Fig. S9a†). The ESI MS of 4 exhibits a prominent ion peak at a mass-to-charge (m/z) ratio of 372.2 (Fig. S9b†), whose mass and isotope distribution pattern correspond to [FeIV(O)(13-TMC)(NCS)]+ (calculated m/z 372.2). When 4 was prepared with isotopically labeled PhI18O in the presence of H218O, the mass peak corresponding to 4 shifted to m/z 374.2 (Fig. S9b,† inset), indicating that 4 contains an oxygen atom. The rRaman spectrum of 4, obtained upon 407 nm excitation in CH3CN at −40 °C, exhibits an isotope-sensitive band at 848 cm−1, which shifted to 810 cm−1 when 4-18O was generated with isotopically labeled PhI18O in the presence of a small amount of H218O in CH3CN (Fig. S9c†). The observed isotopic shift of −38 cm−1 with 18O-substitution is in agreement with the calculated value (Δνcalc = −37 cm−1) for an Fe–O diatomic vibration. Thus, the peak at 848 cm−1 is assigned to the Fe–O stretch, as reported in mononuclear nonheme iron(IV)–oxo complexes bearing 14-TMC and N4Py ligands (N4Py = N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine).15a,c,16 The low-field Mössbauer spectrum of 4 recorded at 4.2 K (Fig. 2c) consists of a quadrupole doublet with δ = 0.07 mm s−1 and ΔEQ = 1.34 mm s−1, characteristic of an S = 1 FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species.16 The S = 1 ground state was confirmed by a high-field experiment (ESI, Fig. S10†). The Mössbauer parameters used in the simulation are listed in ESI, Table S1.† Based on the spectroscopic characterization of 4, we are able to confirm unambiguously that 4 is an iron(IV)–oxo complex binding an anionic axial ligand (NCS−), [FeIV(O)(13-TMC)(NCS)]+. We therefore conclude that addition of an anionic ligand to high-spin iron(III)–alkylperoxo complexes (2) afforded the formation of an iron(IV)–oxo complex (4) (Scheme 2, reaction b) (vide infra).
O species.16 The S = 1 ground state was confirmed by a high-field experiment (ESI, Fig. S10†). The Mössbauer parameters used in the simulation are listed in ESI, Table S1.† Based on the spectroscopic characterization of 4, we are able to confirm unambiguously that 4 is an iron(IV)–oxo complex binding an anionic axial ligand (NCS−), [FeIV(O)(13-TMC)(NCS)]+. We therefore conclude that addition of an anionic ligand to high-spin iron(III)–alkylperoxo complexes (2) afforded the formation of an iron(IV)–oxo complex (4) (Scheme 2, reaction b) (vide infra).
Since the conversion of 2 to 4 was too fast to follow the reaction with a UV-vis spectrophotometer (ESI, Fig. S5†), the reaction of 2 with NCS− was investigated with stopped-flow spectrophotometry. As shown in Fig. 4, addition of NCS− to the solution of BH2 in CH3CN at −40 °C generated a new intermediate 3 with an absorption band at 458 nm within 0.1 s, which was then fully converted to 4 within 40 s. The characterization of the new intermediate 3 was attempted spectroscopically despite its short lifetime. The X-band EPR spectra of 3, which was prepared by reacting BH2 and NCS− in CH3CN at −40 °C and frozen immediately at the given times, exhibit signals corresponding to high-spin iron(III) species (Fig. 4c); the signal intensities were low due to its fast conversion to an EPR inactive iron(IV)–oxo species. It should also be noted that signals corresponding to low-spin iron(III) species were not observed in the EPR measurements (Fig. 4c). The Mössbauer spectra of 3 recorded at 4.2 K in a parallel applied field of 600 G or 7 T are depicted in Fig. 2. The weak field spectrum (Fig. 2d) shows the presence of two minor doublets and a major broad magnetic component. The two doublets are associated with an FeII (15% of total iron, green line) and an FeIVO (20% of total iron, blue line) species as judged from their parameters (ESI, Table S1†). The parameters of the FeIV doublet are identical to those of genuine 4, but those of the FeII doublet (δ = 0.98 mm s−1, ΔEQ = 3.15 mm s−1) are not the same as those of 1, indicating a difference in the iron coordination sphere.18 Such a difference could result from the substitution of a CH3CN ligand for an NCS− ligand. Fig. 2e shows the spectrum recorded at 7 T after subtraction of the FeIVO species. The spectrum is very similar to that of BH2, although the highest velocity line is split in two, revealing the presence of two high-spin ferric species. The spectrum can be fitted with two high-spin FeIII components, one accounting for 13% of total iron with parameters that are common for octahedral high-spin FeIII (orange line), and a majority species (42% of total iron, pink line) with atypical parameters (ESI, Fig. S11 and Table S1†) similar to those of BH2, but with an even larger zero-field splitting value. Owing to its peculiar parameters similar to but distinct from those of BH2, the latter component is assigned to 3, and the former component with classical parameters is assigned to a degradation product.
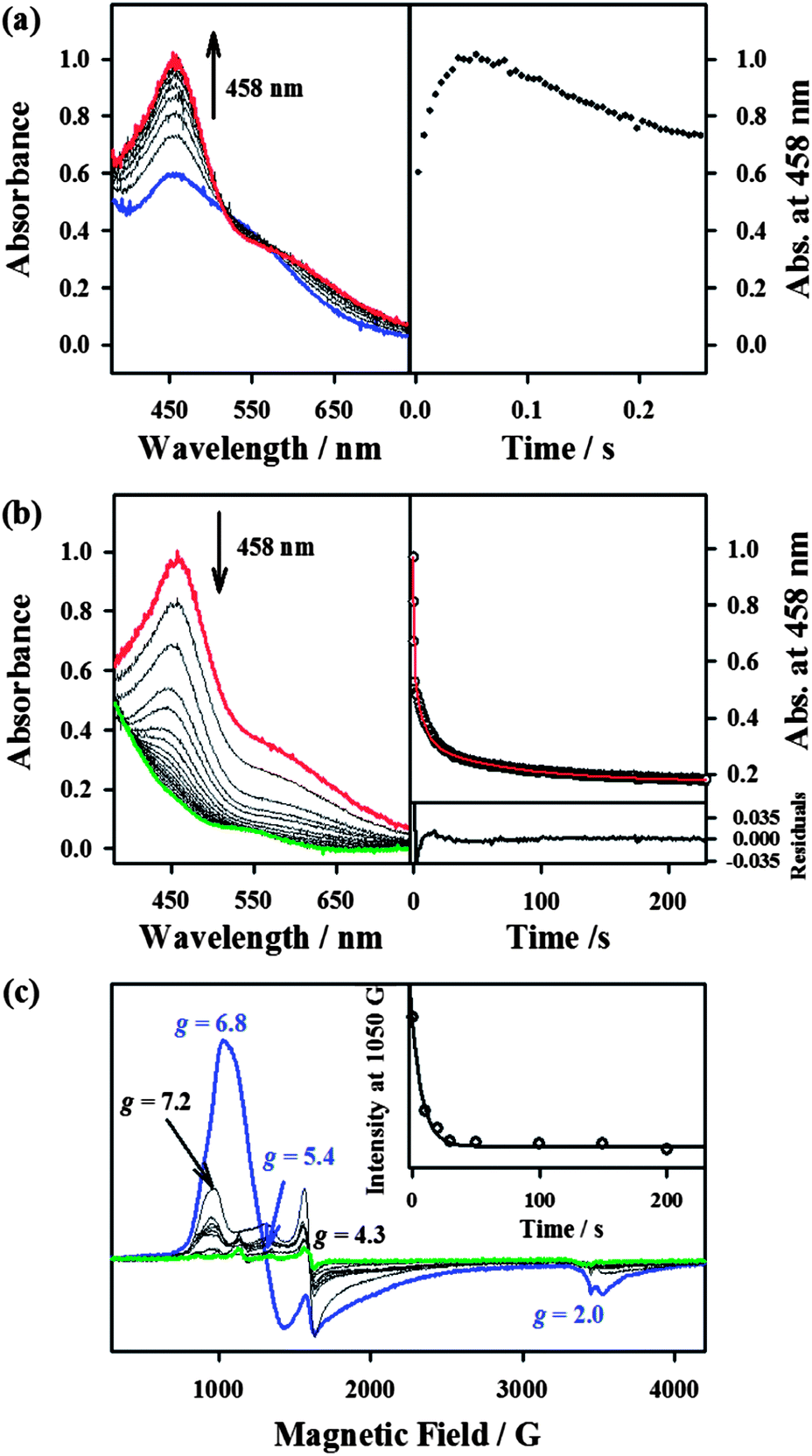 | ||
| Fig. 4 Spectral changes and time courses obtained in the reactions of BH2 (0.50 mM) with thiocyanate (0.60 mM), using single-mixing stopped-flow spectrophotometry and monitoring the formation of the presumed BH2-NCS (3) intermediate at 458 nm in CH3CN at −40 °C. (a) UV-vis spectral changes (left) and time trace (right) recorded during the formation of BH2-NCS (200 ms). (b) UV-vis spectral changes (left) and time trace (upper right) recorded during the decay of BH2-NCS (300 s). (c) X-band EPR spectral changes of BH2 (0.50 mM) (blue) upon the addition of thiocyanate (0.60 mM). The spectra were taken at 0, 10, 20, 30, 50, 100, 150, 200, and 300 s after NCS− was added in CH3CN at −40 °C. BH2-NCS (black) with g = 7.2, 4.3 was formed upon addition of NCS−. The final spectrum is shown with a green line. Inset shows the time dependence of the EPR signal intensity at 1050 G. | ||
The structure of 3 was further corroborated with density functional theory (DFT) calculations.19,20 First, the structure of the DFT-optimized BH2 is shown in Fig. 5a. The energetically lowest structure is in agreement with the high-spin iron(III) state determined in experiments; 9.3 kcal mol−1 lower than the S = 3/2 intermediate-spin state and 16.4 kcal mol−1 lower than the S = 1/2 low-spin state. Addition of NCS− to this structure to form 3 (Fig. 5b) significantly lowers the S = 1/2 low-spin state closer to the ground state on an electron energy level, 0.4 kcal mol−1 higher than the S = 5/2 high-spin state. However, including zero-point vibrational energies (and other correction factors) and looking at the free energy (see ESI, DFT methods description and Table S4†), the S = 5/2 high-spin state is likely to be even more stable than this. Hence, we consider that the DFT-optimized structure of 3 is in agreement with experiments. Based on the EPR and Mössbauer spectroscopic analysis and the DFT-optimized structure, we propose that 3 is a high-spin iron(III)–alkylperoxo complex binding an anionic axial ligand, [FeIII(OOC(CH3)3)(13-TMC)(NCS)]+ (3),21 which is rapidly converted to 4via O–O bond cleavage (vide infra).
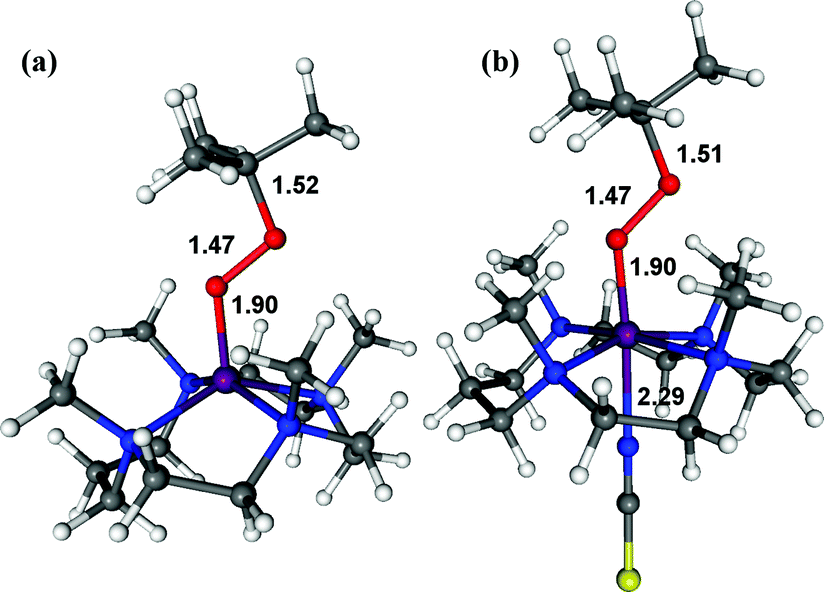 | ||
| Fig. 5 DFT-calculated structures of BH2 (a) and 3 (b). The O–C, O–O, O–Fe, and Fe–NCS bond lengths are indicated in Å (Fe, purple; N, blue; O, red; C, grey; S, yellow; H, white). | ||
Then, how is the O–O bond of the alkylperoxide of 3 cleaved to form 4? Two possible mechanisms are considered, namely O–O bond homolysis (Scheme 2, reaction c) and O–O bond heterolysis followed by a one electron reduction (Scheme 2, reactions d and e). Since cumyl hydroperoxide is a well-known mechanistic probe that can be used to distinguish homolytic vs heterolytic O–O bond cleaving pathways,5b,22 the products formed in the reaction solution were analyzed. The product analysis revealed the exclusive formation of acetophenone (∼80% based on the amount of the intermediate CH2) (Scheme 3, pathway a), as opposed to cumyl alcohol (0%) (Scheme 3, pathway b) (ESI, Experimental section†). The observation of the acetophenone formation as the sole product demonstrates that the conversion of 3 to 4 occurs exclusively via O–O bond homolysis (Scheme 2, reaction c highlighted with a red color), not via O–O bond heterolysis followed by a one electron reduction (Scheme 2, reactions d and e). As a conclusion, we have shown here that the binding of an anionic axial ligand by a high-spin iron(III)–alkylperoxo complex bearing an N-tetramethylated macrocyclic ligand facilitates O–O bond cleavage via the “push effect”, which has been well established in heme enzymes (e.g., CYP 450 and peroxidases) and their model reactions.23,24 We have also shown that O–O bond cleavage of a high-spin iron(III)–alkylperoxo complex binding an anionic axial ligand occurs homolytically, resulting in the generation of its corresponding iron(IV)–oxo complex (Scheme 2, reactions b and c).
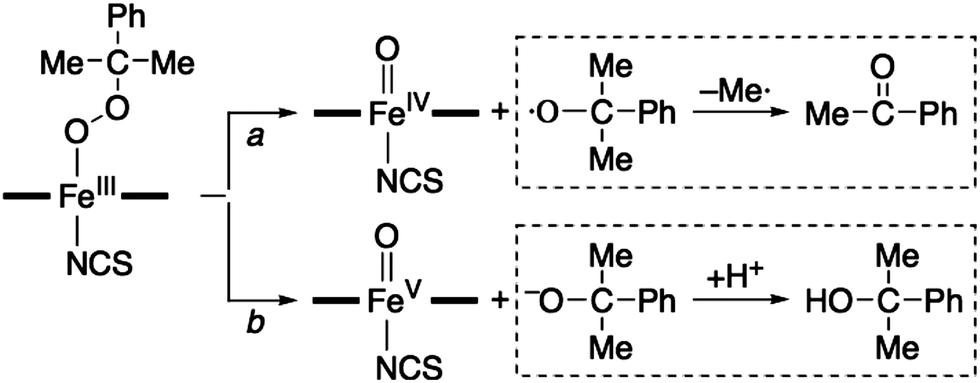 | ||
| Scheme 3 | ||
Conclusion
We have reported the synthesis and characterization of high-spin iron(III)–alkylperoxo complexes each bearing a macrocyclic ligand. The intermediates are stable at low temperature, but are converted to an iron(IV)–oxo complex rapidly upon binding of an anionic axial ligand. We have also provided strong evidence that the conversion of the high-spin iron(III)–alkylperoxo complexes to the iron(IV)–oxo species occurs via O–O bond homolysis. These observations imply that binding of an anionic axial ligand by high-spin iron(III)–alkylperoxo species facilitates O–O bond cleavage, not Fe–O bond cleavage, and that the O–O bond cleavage occurs via a homolytic O–O cleavage mechanism. These results are in sharp contrast to those reported previously which showed that nonheme high-spin iron(III)–alkylperoxo complexes possess weak Fe–O and strong O–O bonds that give rise to Fe–O bond homolysis rather than O–O bond cleavage.8,9a,10b Further, it should be noted that the present results are different from the previously proposed role of the thiolate axial ligand trans to the iron-hydroperoxo moiety in SOR and its model compounds, i.e. that the electron-donating thiolate ligand increases the lifetime of the high-spin iron(III)–hydroperoxo species to allow its protonation and subsequent release of H2O2 in SOR.7a,11d,25 Further mechanistic investigation, including DFT calculations, is underway in this laboratory to understand the factors (e.g., effects of the axial ligand and the H.S. and L.S. iron(III) spin states) that affect the activation of iron(III)–alkylperoxo and –hydroperoxo complexes in nonheme iron models.26Acknowledgements
The research at EWU was supported by NRF/MEST of Korea through the CRI (2-2012-1794-001-1 to W.N.), GRL (2010-00353 to W.N.), and Basic Research Program (2010-0002558 to M.S.S.). J.M.L. acknowledges the support of the Region Rhone-Alpes through contract CIBLE 07 016335 and Labex ARCANE (ANR-11-LABX-0003-01). T.O. thanks the Ministry of Education, Culture, Sports, Science and Technology of Japan through the Global COE program and Priority Area (no. 22018026).Notes and references
- W. Nam, Acc. Chem. Res., 2007, 40, 465 CrossRef CAS , and review articles in the special issue.
- (a) P. R. Ortiz de Montellano in Cytochrome P450: Structure, Mechanism, and Biochemistry, Kluwer Academic/Plenum Publishers, New York, 2005 Search PubMed; (b) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed; (c) S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS PubMed; (d) J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
- (a) E. G. Kovaleva and J. D. Lipscomb, Nat. Chem. Biol., 2008, 4, 186–193 CrossRef CAS PubMed; (b) E. I. Solomon, S. D. Wong, L. V. Liu, A. Decker and M. S. Chow, Curr. Opin. Chem. Biol., 2009, 13, 99–113 CrossRef CAS PubMed.
- (a) J. Cho, S. Jeon, S. A. Wilson, L. V. Liu, E. A. Kang, J. J. Braymer, M. H. Lim, B. Hedman, K. O. Hodgson, J. S. Valentine, E. I. Solomon and W. Nam, Nature, 2011, 478, 502–505 CrossRef CAS PubMed; (b) J. Kaizer, M. Costas and L. Que, Jr, Angew. Chem., Int. Ed., 2003, 42, 3671–3673 CrossRef CAS PubMed; (c) M. S. Seo, T. Kamachi, T. Kouno, K. Murata, M. J. Park, K. Yoshizawa and W. Nam, Angew. Chem., Int. Ed., 2007, 46, 2291–2294 CrossRef CAS PubMed; (d) M. P. Jensen, A. M. i Payeras, A. T. Fiedler, M. Costas, J. Kaizer, A. Stubna, E. Münck and L. Que, Jr, Inorg. Chem., 2007, 46, 2398–2408 CrossRef CAS PubMed.
- (a) M. K. Coggins and J. A. Kovacs, J. Am. Chem. Soc., 2011, 133, 12470–12473 CrossRef CAS PubMed; (b) M. K. Coggins, V. Martin-Diaconescu, S. DeBeer and J. A. Kovacs, J. Am. Chem. Soc., 2013, 135, 4260–4272 CrossRef CAS PubMed.
- (a) F. E. Jenney, Jr, M. F. J. M. Verhagen, X. Cui and M. W. W. Adams, Science, 1999, 286, 306–309 CrossRef; (b) D. A. Kurtz, Jr, Acc. Chem. Res., 2004, 37, 902–908 CrossRef PubMed.
- (a) J. A. Kovacs, Chem. Rev., 2004, 104, 825–848 CrossRef CAS PubMed; (b) J. A. Kovacs and L. M. Brines, Acc. Chem. Res., 2007, 40, 501–509 CrossRef CAS PubMed; (c) J. Shearer, R. C. Scarrow and J. A. Kovacs, J. Am. Chem. Soc., 2002, 124, 11709–11717 CrossRef CAS PubMed; (d) T. Kitagawa, A. Dey, P. Lugo-Mas, J. B. Benedict, W. Kaminsky, E. I. Solomon and J. A. Kovacs, J. Am. Chem. Soc., 2006, 128, 14448–14449 CrossRef CAS PubMed; (e) E. Nam, P. E. Alokolaro, R. D. Swartz, M. C. Gleaves, J. Pikul and J. A. Kovacs, Inorg. Chem., 2011, 50, 1592–1602 CrossRef CAS PubMed.
- (a) D. Krishnamurthy, G. D. Kasper, F. Namuswe, W. D. Kerber, A. A. N. Sarjeant, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2006, 128, 14222–14223 CrossRef CAS PubMed; (b) F. Namuswe, G. D. Kasper, A. A. N. Sarjeant, T. Hayashi, C. M. Krest, M. T. Green, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2008, 130, 14189–14200 CrossRef CAS PubMed; (c) F. Namuswe, T. Hayashi, Y. Jiang, G. D. Kasper, A. A. N. Sarjeant, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2010, 132, 157–167 CrossRef CAS PubMed.
- (a) A. Dey and E. I. Solomon, Inorg. Chim. Acta, 2010, 363, 2762–2767 CrossRef CAS PubMed; (b) A. Franke, C. Feritinger and R. van Eldik, Chem.–Eur. J., 2012, 18, 6935–6949 CrossRef CAS PubMed.
- (a) N. Lehnert, R. Y. N. Ho, L. Que, Jr and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 8271–8290 CrossRef CAS PubMed; (b) N. Lehnert, R. Y. N. Ho, L. Que, Jr and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 12802–12816 CrossRef CAS PubMed.
- (a) Y. Zang, J. Kim, Y. Dong, E. C. Wilkinson, E. H. Appelman and L. Que, Jr, J. Am. Chem. Soc., 1997, 119, 4197–4205 CrossRef CAS; (b) A. Wada, S. Ogo, Y. Watanabe, M. Mukai, T. Kitagawa, K. Jitsukawa, H. Masuda and H. Einaga, Inorg. Chem., 1999, 38, 3592–3593 CrossRef CAS PubMed; (c) A. Wada, S. Ogo, S. Nagatomo, T. Kitagawa, Y. Watanabe, K. Jitsukawa and H. Masuda, Inorg. Chem., 2002, 41, 616–618 CrossRef CAS PubMed; (d) M. R. Bukowski, H. L. Halfen, T. A. van den Berg, J. A. Halfen and L. Que, Jr, Angew. Chem., Int. Ed., 2005, 44, 584–587 CrossRef CAS PubMed; (e) J. Bautz, P. Comba and L. Que, Jr, Inorg. Chem., 2006, 45, 7077–7082 CrossRef CAS PubMed; (f) X. Shan, J.-U. Rohde, K. D. Koehntop, Y. Zhou, M. R. Bukowski, M. Costas, K. Fujisawa and L. Que, Jr, Inorg. Chem., 2007, 46, 8410–8417 CrossRef CAS PubMed; (g) S. Gosiewska, H. P. Permentier, A. P. Bruins, G. van Koten and R. J. M. Klein Gebbink, Dalton Trans., 2007, 3365–3368 RSC.
- (a) E. K. Barefield, Coord. Chem. Rev., 2010, 254, 1607–1627 CrossRef; (b) J. Cho, R. Sarangi and W. Nam, Acc. Chem. Res., 2012, 45, 1321–1330 CrossRef CAS PubMed.
- S. Hong, H. So, H. Yoon, K.-B. Cho, Y.-M. Lee, S. Fukuzumi and W. Nam, Dalton Trans., 2013, 42, 7842–7845 RSC.
- J.-J. Girerd, F. Banse and A. J. Simaan, Struct. Bonding, 2000, 97, 145–177 CrossRef CAS.
- (a) C. V. Sastri, M. J. Park, T. Ohta, T. A. Jackson, A. Stubna, M. S. Seo, J. Lee, J. Kim, T. Kitagawa, E. Münck, L. Que, Jr and W. Nam, J. Am. Chem. Soc., 2005, 127, 12494–12495 CrossRef CAS PubMed; (b) C. V. Sastri, J. Lee, K. Oh, Y. J. Lee, J. Lee, T. A. Jackson, K. Ray, H. Hirao, W. Shin, J. A. Halfen, J. Kim, L. Que, Jr, S. Shaik and W. Nam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19181–19186 CrossRef CAS PubMed; (c) T. A. Jackson, J.-U. Rohde, M. S. Seo, C. V. Sastri, R. DeHont, A. Stubna, T. Ohta, T. Kitagawa, E. Münck, W. Nam and L. Que, Jr, J. Am. Chem. Soc., 2008, 130, 12394–12407 CrossRef CAS PubMed; (d) S. A. Wilson, J. Chen, S. Hong, Y.-M. Lee, M. Clemancey, R. Garcia-Serres, T. Nomura, T. Ogura, J.-M. Latour, B. Hedman, K. O. Hodgson, W. Nam and E. I. Solomon, J. Am. Chem. Soc., 2012, 134, 11791–11806 CrossRef CAS PubMed.
- (a) S. P. de Visser, J.-U. Rohde, Y.-M. Lee, J. Cho and W. Nam, Coord. Chem. Rev., 2013, 257, 381–393 CrossRef CAS; (b) A. R. McDonald and L. Que, Jr, Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS; (c) W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed.
- [FeII(13-TMC)(NCS)](CF3SO3) was synthesized as an authentic compound and its spectroscopic and structural characterizations were carried out (see ESI,† Fig. S7 for ESI MS and EPR spectra and Fig. S8 and Tables S2 and S3). X-ray crystal structure of [FeII(13-TMC)(NCS)](CF3SO3) clearly indicates that the N-donor of thiocyanate is coordinated to the FeII centre.
- This species is identified as [FeII(13-TMC)(NCS)]+ based on the ESI MS measurement of the reaction solution. The ESI MS clearly shows the presence of [FeII(13-TMC)(NCS)]+, not 1 (data not shown).
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- DFT optimized structures were obtained based on the X-ray crystal structure of [FeII(13-TMC)(NCS)](CF3SO3) (see ref. 17).
- (a) An octahedral iron(III) complex bearing an N-tetrametylated cyclam ligand is shown to contain high-spin Fe(III) (S = 5/2): J. F. Berry, E. Bill, R. Garcia-Serres, F. Neese, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2006, 45, 2027–2037 CrossRef CAS PubMed; (b) Our DFT calculations indicate that the NCS− binding is endothermic by 10.8 kcal mol−1, providing an explanation for its instability.
- (a) D. V. Avila, C. E. Brown, K. U. Ingold and J. Lusztyk, J. Am. Chem. Soc., 1993, 115, 466–470 CrossRef CAS; (b) M. Costas, K. Chen and L. Que, Jr, Coord. Chem. Rev., 2000, 200–202, 517–544 CrossRef CAS; (c) E. Baciocchi, M. Bietti, M. Salamone and S. Stteenken, J. Org. Chem., 2002, 67, 2266–2270 CrossRef CAS PubMed; (d) T. Tano, H. Sugimoto, N. Fujieda and S. Itoh, Eur. J. Inorg. Chem., 2012, 4099–4103 CrossRef CAS.
- (a) S. Yoshioka, S. Takahashi, K. Ishimori and I. Morishima, J. Inorg. Biochem., 2000, 81, 141–151 CrossRef CAS PubMed; (b) D. L. Harris and G. H. Loew, J. Am. Chem. Soc., 1998, 120, 8941–8948 CrossRef CAS; (c) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2887 CrossRef CAS PubMed; (d) T. L. Poulos, J. Biol. Inorg. Chem., 1996, 1, 356–359 CrossRef CAS; (e) D. B. Goodin, J. Biol. Inorg. Chem., 1996, 1, 360–363 CrossRef CAS; (f) J. H. Dawson, Science, 1988, 240, 433–439 CAS.
- (a) N. Hessenauer-Ilicheva, A. Franke, D. Meyer, W.-D. Woggon and R. van eldik, Chem.–Eur. J., 2009, 15, 2941–2959 CrossRef CAS PubMed; (b) W. Nam, H. J. Han, S.-Y. Oh, Y. J. Lee, M.-H. Choi, S.-Y. Han, C. Kim, S. K. Woo and W. Shin, J. Am. Chem. Soc., 2000, 122, 8677–8684 CrossRef CAS; (c) T. Matsui, S. Ozaki and Y. Watanabe, J. Am. Chem. Soc., 1999, 121, 9952–9957 CrossRef CAS; (d) W. Nam, H. J. Choi, H. J. Han, S. H. Cho, H. J. Lee and S.-Y. Han, Chem. Commun., 1999, 387–388 RSC; (e) F. Minisci, F. Fontana, S. Araneo, F. Recupero, S. Banfi and S. Quici, J. Am. Chem. Soc., 1995, 117, 226–232 CrossRef CAS.
- (a) M. D. Clay, F. E. Jenney, Jr, P. L. Hagedoorn, G. N. George, M. W. W. Adams and M. K. Johnson, J. Am. Chem. Soc., 2002, 124, 788–805 CrossRef CAS PubMed; (b) R. Silaghi-Dumitrescu, I. Silaghi-Dumitrescu, E. D. Coulter and D. M. Kurtz, Jr, Inorg. Chem., 2003, 42, 446–456 CrossRef CAS PubMed.
- L. V. Liu, S. Hong, J. Cho, W. Nam and E. I. Solomon, J. Am. Chem. Soc., 2013, 135, 3286–3299 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section; synthesis, characterization, kinetics and DFT details. CCDC reference number 959085. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc52236a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |