 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long-lived metal complexes open up microsecond lifetime imaging microscopy under multiphoton excitation: from FLIM to PLIM and beyond†
Elizabeth
Baggaley
a,
Stanley W.
Botchway
*b,
John W.
Haycock
*c,
Hayley
Morris
c,
Igor V.
Sazanovich
a,
J. A. Gareth
Williams
*d and
Julia A.
Weinstein
*a
aDepartment of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. E-mail: julia.weinstein@sheffield.ac.uk
bCentral Laser Facility, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Oxfordshire, OX11 0QX, UK. E-mail: Stan.Botchway@stfc.ac.uk
cDepartment of Materials Science and Engineering, The Kroto Research Institute, University of Sheffield, Sheffield, S3 7HQ, UK. E-mail: j.w.haycock@sheffield.ac.uk
dDepartment of Chemistry, University of Durham, Durham, DH1 3LE, UK. E-mail: j.a.g.williams@durham.ac.uk
First published on 16th October 2013
Abstract
Lifetime imaging microscopy with sub-micron resolution provides essential understanding of living systems by allowing both the visualisation of their structure, and the sensing of bio-relevant analytes in vivo using external probes. Chemistry is pivotal for the development of the next generation of bio-tools, where contrast, sensitivity, and molecular specificity facilitate observation of processes fundamental to life. A fundamental limitation at present is the nanosecond lifetime of conventional fluorescent probes which typically confines the sensitivity to sub-nanosecond changes, whilst nanosecond background autofluorescence compromises the contrast. High-resolution visualization with complete background rejection and simultaneous mapping of bio-relevant analytes including oxygen – with sensitivity orders of magnitude higher than that currently attainable – can be achieved using time-resolved emission imaging microscopy (TREM) in conjunction with probes with microsecond (or longer) lifetimes. Yet the microsecond timescale has so far been incompatible with available multiphoton excitation/detection technologies. Here we realize for the first time microsecond-imaging with multiphoton excitation whilst maintaining the essential sub-micron spatial resolution. The new method is background-free and expands available imaging and sensing timescales 1000-fold. Exploiting the first engineered water-soluble member of a family of remarkably emissive platinum-based, microsecond-lived probes amongst others, we demonstrate (i) the first instance of background-free multiphoton-excited microsecond depth imaging of live cells and histological tissues, (ii) over an order-of-magnitude variation in the probe lifetime in vivo in response to the local microenvironment. The concept of two-photon TREM can be seen as “FLIM + PLIM” as it can be used on any timescale, from ultrafast fluorescence of organic molecules to slower emission of transition metal complexes or lanthanides/actinides, and combinations thereof. It brings together transition metal complexes as versatile emissive probes with the new multiphoton-excitation/microsecond-detection approach to create a transformative framework for multiphoton imaging and sensing across biological, medicinal and material sciences.
Introduction
Emission imaging microscopy1–4 has revolutionized our understanding of living systems by allowing their structure to be vizualised with sub-micron resolution and biochemical function to be monitored at the molecular level. Real-time fluorescence imaging relies on biocompatible labels to “light up” targeted intracellular structures, or to report on their biochemical microenvironment through a change in the emission response5–7 using signal intensity, wavelength, or lifetime as observables. The vast majority of labels to date are fluorescent GFP-type proteins,6a,b or synthetic organic molecules which fluoresce in the visible range, with a lifetime of a few nanoseconds.6cContemporary fluorescence lifetime imaging microscopy (FLIM)8–10 is fundamentally restricted by the nanosecond lifetime of the available probes, which limits both contrast and sensitivity. The sensitivity to small-molecule analytes, including detection of those in vivo, is confined to sub-nanosecond changes, limiting the detection limit of many analytes over physiologically relevant concentration ranges. The contrast is greatly compromised by autofluorescence of endogenous biomolecules, which has the same characteristics as most of the currently used exogenous labels, appearing in the visible range with a lifetime of several nanoseconds. Autofluorescence, which progressively increases in intensity from single cells to more complex samples such as tissues or whole organisms, remains a major problem in imaging,11 as no method of absolute autofluorescence rejection presently exists. High-resolution visualization with complete background rejection, could be achieved by employing molecular probes that emit on a substantially longer, microsecond, timescale.15–20 Moreover, at the same time, an orders-of-magnitude increase in sensitivity of lifetime imaging to bio-relevant analytes including oxygen12–14 should be achievable through such an extension in lifetime. This concept of time-resolved emission imaging microscopy (TREM) would allow gating out of autofluorescence at early times and selective imaging of the probe's emission at later times.
Appropriately long emission lifetimes can be obtained by exploiting the formally forbidden process of phosphorescence. Transition metal complexes are ideal candidates for this task, as they may phosphoresce intensely on the microsecond timescale owing to the high spin–orbit coupling associated with the heavy metal ion. This property underpins the success of transition metal complexes as sensors for diverse bio-relevant molecules in vitro.21 The past five years have seen a rapid development of phosphorescent transition metal complexes in biological imaging22–28 yet so far primarily in the steady-state regime. The growth in reported new metal-based labels demonstrates the need to develop methods alternative29 to traditional steady-state or nanosecond imaging for life sciences.
We recently implemented the extremely attractive concept of TREM, by developing a family of remarkably emissive cyclometallated platinum(II) complexes (PtLnCl, Fig. 1a), which have high quantum yields (>70%), emission lifetimes of microseconds, and which are cell-permeable and surprisingly photostable.30 Using these compounds, gated imaging under single-photon excitation was achieved albeit with low spatial resolution.31
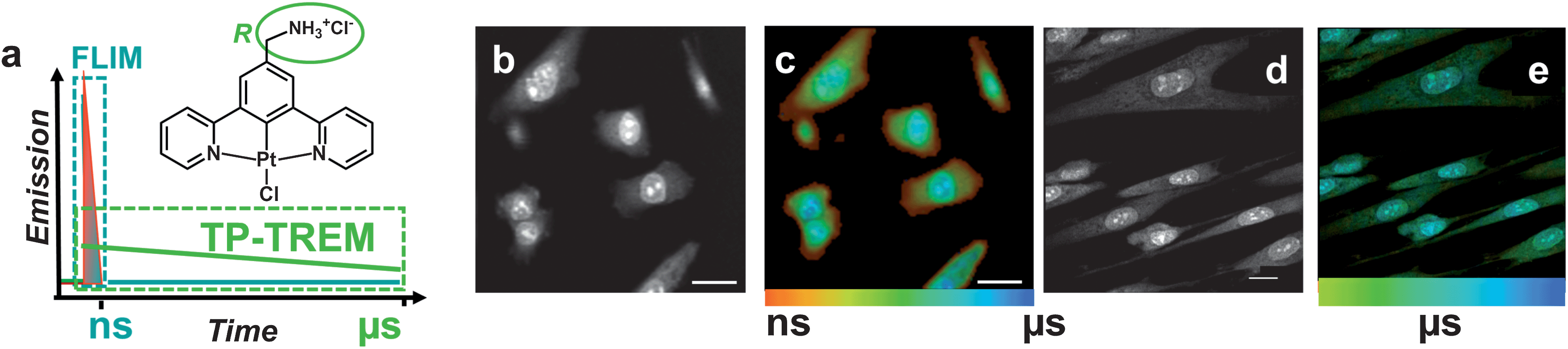 | ||
| Fig. 1 Microsecond multiphoton time-resolved imaging. (a) The concept of the method and the general structure of PtLnCl emissive labels: n = 1, R = H; n = 2, R = CH3; n = 3, R = –CH2–NH3+Cl−. The structure is shown for the water-soluble derivative n = 3. (b and c) Live CHO-K1 cells labelled with PtL1Cl (100 μM, 5 min, 37 °C) (b) TP-TREM intensity image under 760 nm two-photon excitation, reconstructed by integrating total emission intensity pixel-per-pixel; (c) lifetime distribution map corresponding to image (b). (d and e) Live HDF cells incubated with a 100 μM solution of water-soluble PtL3Cl in PBS, 5 min at 37 °C. (d) Intensity image obtained by collecting all decay photos in one channel; (e) lifetime distribution map corresponding to image (d), showing the platinum-based emission component on the time scale 2–10 μs. Scale bars 20 μm. | ||
However, up to now, it has not been possible to combine microsecond detection with multiphoton excitation and sub-micron resolution in imaging. The incompatibility of microsecond probes with the available high resolution multiphoton excitation/detection technologies arises from the requisite high photon flux that necessitates the use of femtosecond lasers with high repetition rates of ∼100 MHz, whilst the long lifetime of the probe would require low repetition-rate lasers and unsustainably long accumulation times. Thus, current lifetime imaging technology is limited either to short emission lifetimes, but with high spatial resolution; or to much longer and analytically valuable emission lifetimes, but with low spatial resolution.
Here we introduce a significant step forward in the interrogation and understanding of complex and evolving objects by developing and implementing a powerful new method of “two-photon time-resolved emission imaging microscopy” (TP-TREM). The general method overcomes the limitations given above and combines for the first time microsecond imaging with enhanced multiphoton resolution and diffraction-limited point-scanning in a fast correlated individual-photon tagging implementation. It uses phosphorescent rather than fluorescent probes and achieves time-resolved detection on a timescale several orders of magnitude longer than that available in FLIM yet maintaining the essential sub-micron spatial resolution afforded by two-photon excitation, thereby offering dramatic improvements in both contrast and sensitivity in emission imaging.
Exploiting, amongst others, the first water-soluble member of a family of platinum-based oxygen-sensitive probes with microsecond timescale emission, we demonstrate how TP-TREM can provide background-free multiphoton microsecond depth-imaging of diverse live cells and histological tissues, and variation in the probe lifetime in vivo orders of magnitude higher than that available with current techniques or probes.
Results and discussion
The multiphoton microsecond imaging method TP-TREM (Fig. 1) produces a 6D emission surface (x–y–z–I–λ–τ), where each pixel (x–y–z) is characterized by emission decay (τ) from pico- to microseconds, intensity I, and spectral information (λ); high spatial resolution is achieved by two-photon excitation. Integrating the emission intensity at every pixel over the interval of time between the pulses produces the corresponding intensity map. The emission decay kinetics accumulated at each pixel generate a lifetime map in the region of interest, where the lifetime distribution is colour-coded from red (short lifetimes) to blue (long lifetimes), Fig. 1c. Two-photon excitation requires femtosecond lasers with high photon flux,32 usually operating at repetition rates of 80–100 MHz with a time interval of ∼10 ns between pulses, incompatible with microsecond probes.We describe how this intrinsic contradiction has been overcome (see Experimental section for details of instrumentation), as well as how the detection timescales have been extended from the usual 10 ns to microseconds. Pixel-specific emission spectra have been collected to confirm the identity of the emissive species.
We introduce the first water-soluble representative of the family of PtLnCl probes (Fig. 1, n = 3 R = –CH2NH3+Cl−) which allows the probe to be administered in a biocompatible medium without the need for pre-solubilisation in organic solvents that may be harmful to mammalian cells. PtL3Cl absorbs visible light intensely under either one-photon (εmax ∼ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) or two-photon excitation (two-photon absorption cross-section ∼3.5 GM at 760 nm), and does not photobleach under intense prolonged irradiation, similar to other representatives of its family.30,31 The highly structured emission spectra of such complexes30,31 are characteristic of the intraligand 3π–π* transition, thus demonstrating efficient access to the triplet manifold, and its subsequent radiative decay, essential for a long emission lifetime at ambient temperature. The lifetime increases from 170 ns in aerated to 5.4 μs in degassed solutions – indicating the potential for oxygen sensing. All PtLnCl derivatives accumulate within a wide range of live eukaryotic cells within a short incubation time of 5 min, suggesting diffusion-controlled accumulation without an active uptake mechanism, and they retain their impressive emission characteristics within the cell.
000 M−1 cm−1) or two-photon excitation (two-photon absorption cross-section ∼3.5 GM at 760 nm), and does not photobleach under intense prolonged irradiation, similar to other representatives of its family.30,31 The highly structured emission spectra of such complexes30,31 are characteristic of the intraligand 3π–π* transition, thus demonstrating efficient access to the triplet manifold, and its subsequent radiative decay, essential for a long emission lifetime at ambient temperature. The lifetime increases from 170 ns in aerated to 5.4 μs in degassed solutions – indicating the potential for oxygen sensing. All PtLnCl derivatives accumulate within a wide range of live eukaryotic cells within a short incubation time of 5 min, suggesting diffusion-controlled accumulation without an active uptake mechanism, and they retain their impressive emission characteristics within the cell.
The power of the TP-TREM method enabled by the Pt(II)-probes has been demonstrated in recording background-free high-contrast images of live cells and autofluorescent tissues of different morphologies with multiphoton (sub-micron) resolution, as well as simultaneous visualisation of multiple-colour stained samples, and mapping of the intracellular concentration of small molecular analytes exemplified by oxygen.
Live cells: lifetime maps, imaging and sensing
A representative experiment demonstrates a TP-TREM investigation of live Chinese Hamster Ovary (CHO) cells incubated with PtL1Cl (Fig. 1b and c) and of Human Dermal Fibroblast (HDF) cells incubated with PtL3Cl (Fig. 1d and e). The intensity distribution (Fig. 1b and d) of PtLnCl emission under TP-TREM obtained in the broad field of view by integrating intensity pixel-per-pixel demonstrates the preferential localisation of PtLnCl in the nucleus and, especially, nucleoli, with considerably weaker staining of the cytoplasm. The emission spectra collected from different intracellular locations under two-photon excitation (Fig. 2f) are identical to one another, and to that observed in solution under one- or two-photon excitation. The observed vibrational progression characteristic of the 3π–π* emission of the [Pt(Ln)] unit confirms that the emissive moiety remains intact inside the cell.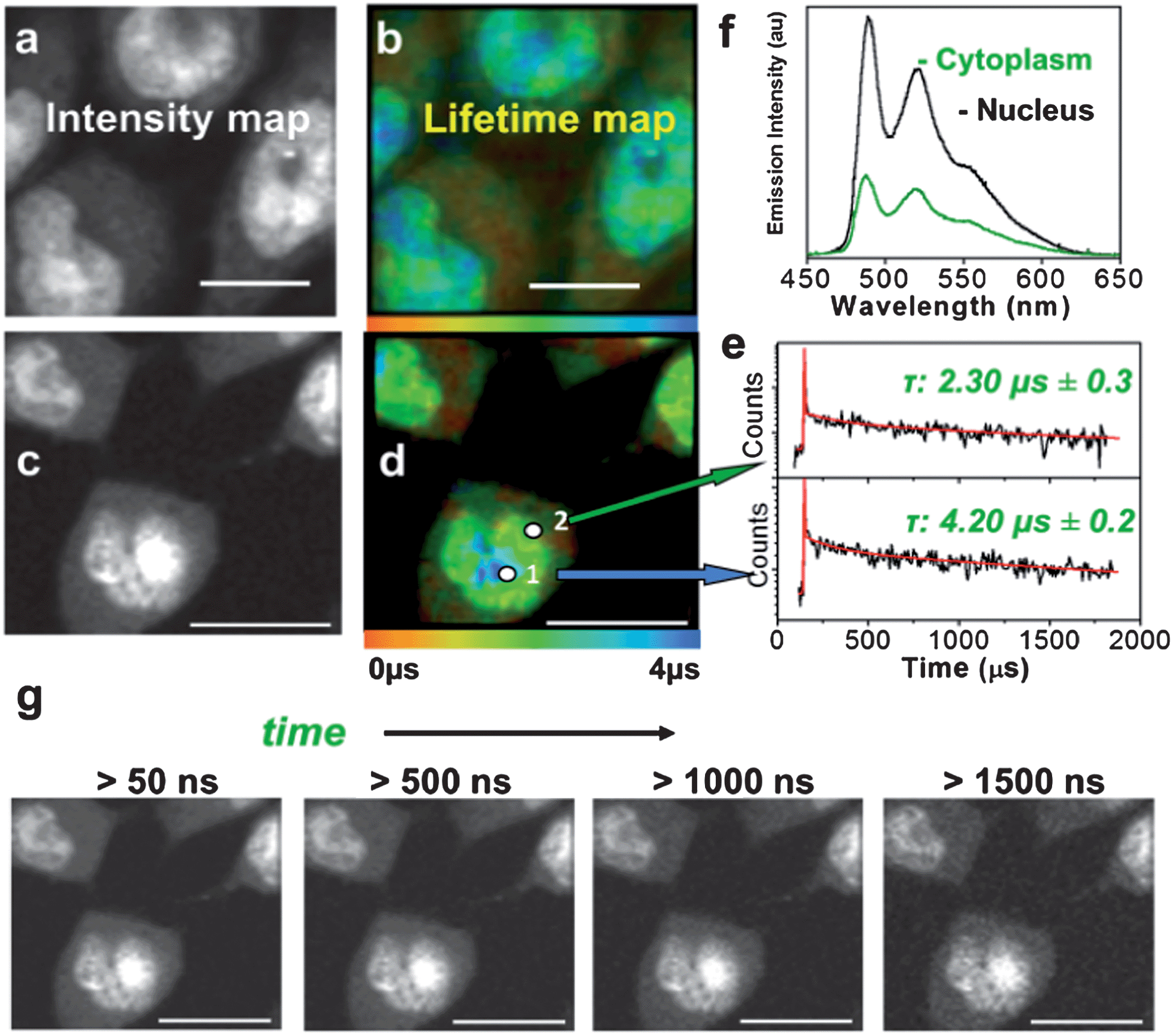 | ||
| Fig. 2 Microsecond imaging of live cells and sensitivity to the microenvironment. TP-TREM of CHO-K1 cells labeled with PtL3Cl (a and b) and PtL1Cl (c–g) under 760 nm, ∼120 fs two-photon excitation. (a and c) Intensity images reconstructed by integrating total emission intensity pixel-per-pixel; (b) lifetime map corresponding to (a); (d) lifetime map corresponding to (c); (e) kinetic traces from pixels 1 and 2 on (d), red line corresponds to the fit with the lifetime of the main component stated [pixel symbol magnified for clarity]; (f) emission spectra from nucleus and cytoplasm on (d); (g) intensity images at progressively longer time delays after the laser pulse for the PtL1Cl labeled cells shown in (c and d). Scale bars 20 μm. See Fig. S6† for steady state transmission and confocal emission image, Fig. S7† for spectra from (a and b). | ||
The lifetime maps reveal a wealth of detail within the cell, illuminating, in particular, nuclear and cell membranes which are not visible in the intensity image. Fig. 1c highlights the general trend of increasing lifetimes when moving from the cell membrane, through the cytoplasm and into the nucleolus. The lifetime map obtained with high spatial resolution for CHO-K1 cells incubated with the water-soluble PtL3Cl (Fig. 2b) or PtL1Cl (Fig. 2d) quantifies the marked influence of the localisation and microenvironment on the emission lifetime of the label. The emission lifetime of PtLnCl varies by a factor of >20 depending on the location: e.g., for PtL1Cl, from ∼300 ns in the membrane, to 2.3 ± 0.3 μs in the cytoplasm, and to 4.2 ± 0.2 μs in the nucleus (Fig. 2e). Moreover, discrete zones within the nucleus have particularly long lifetimes of ∼ 5.8 μs, close to that in fully deoxygenated solution (deep blue areas, Fig. 2b and d). Such a difference could arise due to the variable degree of protection from quenching by oxygen, or to the differing extent of self-quenching via interactions with another PtLnCl molecule. Binding to proteins or intercalating into DNA may also offer such variable protection. Interaction with DNA was confirmed by titration studies,31 whilst cluster-like Pt-labelled structures within the nuclei are also consistent with intracellular binding to DNA and possibly to RNA.33
The influence of oxygen is of particular interest: the emission of PtLnCl compounds in solution is strongly quenched by oxygen with a Stern–Volmer constant of 108 to 109 M−1 s−1,30 leading to a 15 to 20-fold change in its emission lifetime, from 5–7 μs in a fully deoxygenated to 170–500 ns in an aerated solution; the same values were obtained by TP-TREM in solution in a gas chamber on the microscope. It is therefore likely that the observed significant variation in the lifetime is reflecting, at least in part, the oxygen concentration in a particular location within the cell. This notion is supported by a significant increase in the emission lifetime of the Pt-labels in the cytoplasm when equilibrated in an atmosphere of 95%N2/5% CO2, for instance, from 1.49 μs to 6 μs for PtL3Cl-labelled CHO cells. The probes can comfortably sense oxygen to sub-micromolar level; slightly longer-lived probes would allow nanomolar-range detection. In contrast to the long-lived emission of Pt-labels, the short-lived emission of organic labels is almost unaffected by oxygen. The ∼2 ns lifetime of a fluorescent label would only be reduced by ∼0.2%, to 1.996 ns, under deoxygenated vs. aerated conditions at typical oxygen concentrations of 100 μM, even assuming a maximum possible diffusion-controlled quenching rate constant of ∼1010 M−1 s−1. Thus the use of ∼1000 times longer emission lifetimes of the Pt-probes in comparison to fluorescent labels leads to a greatly increased sensitivity to the local environment in TP-TREM compared to fluorescent lifetime imaging. The time-gated images accumulated at progressively longer delays after the ∼120 fs excitation pulse demonstrate that even at a strikingly long delay of 1.5 μs – after which time, neither autofluorescence nor any organic fluorescent label would retain any intensity – the subcellular structures are still clearly visualised (Fig. 2g).
Of particular relevance to imaging objects undergoing temporal evolution (such as dividing cells or self-assembly), is the achievement of simultaneous visualisation of a multitude of subcellular structures employing a single label in a single experiment. The 3D-imaging is demonstrated by a Z-scan projection of PtLnCl-labelled CHO cells, where the total intensity for each slice is obtained by binning all emission decay photons per pixel (ESI Video†).
Application of TP-TREM to a variety of phenotypically different eukaryotic cell types, including human cervical cancer (HeLa), human melanoma (HBL), human keratinocyte (HaCat), human dermal fibroblast (HDF) and rat Schwann cells (RN22), yielded the same type of information as discussed above on the example of CHO cells, demonstrating the wide general applicability of the new approach.
Background-free microsecond imaging of tissues
Autofluorescence is particularly forbidding in tissue imaging due to the intrinsically highly fluorescent nature of many extracellular matrix proteins. Two-photon TREM imaging has been performed using PtLnCl-labelled multicellular tissue samples of rat skin, peripheral nerve and liver, representative of considerably different tissue morphologies, (Fig. 3, 4 and S8–S10†). The strong autofluorescence at the epidermis, predominantly due to a high local concentration of keratin, dominates the intensity-based image of a section of unlabelled skin, where basic but poorly-defined morphology of the stratum corneum and supra-basal keratinocytes can be identified (Fig. 3a). Likewise, unlabelled liver tissue sections autofluoresce strongly due to the presence of matrix protein collagen (Fig. S9†). The intensity-based image of the tissue sections labelled with PtL1Cl shows an almost uniform distribution of emission intensity (Fig. 3b, left – skin; Fig. 4a – liver) where no substructures can be identified with confidence. In contrast, multiphoton images of PtL1Cl-labelled tissues (Fig. 3, or PtL3Cl-labelled, Fig. S12†) recorded after a time delay of >50 ns reveal intact cellular structures. Keratinocytes and some weak staining of the hyperkeratinised, terminally differentiated keratinocytes, and of the associated extracellular matrix, are observed for skin (Fig. 3b and S12†). Staining of the nucleus of the hepatocyte cells is observed for liver (Fig. 4b). The nuclear localisation of PtL1Cl was confirmed by co-labelling with the nuclear stain DAPI (Fig. S8 and S10†). Emission spectra obtained from the nuclei of both keratinocyte and hepatocyte cells under two-photon excitation are characteristic of the [Pt(Ln)] moiety (skin, Fig. 3c; liver, Fig. S11†).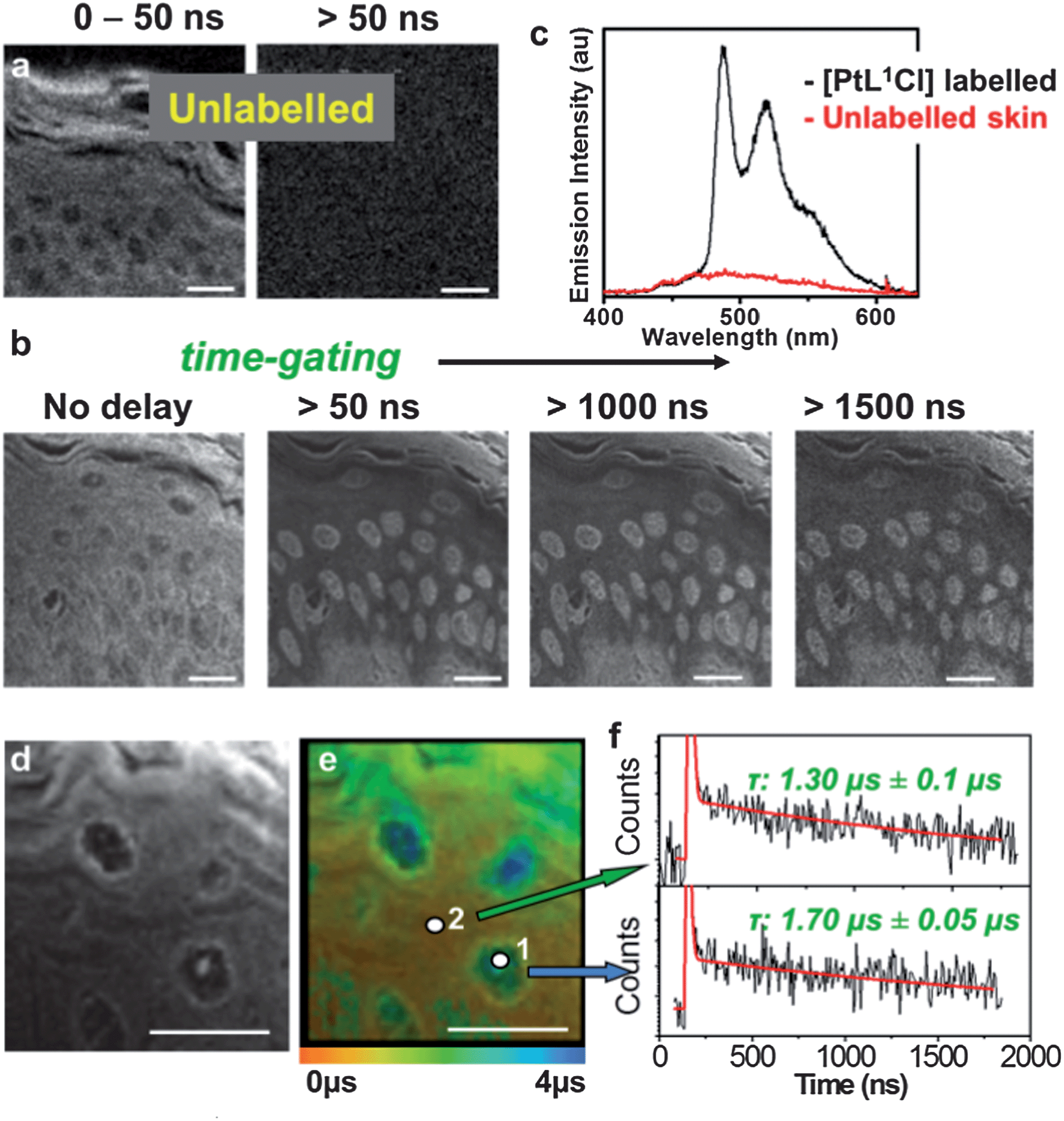 | ||
| Fig. 3 PtL1Cl-labelled skin tissue section. (a) Intensity image of unlabelled skin epidermis at early times (0–50 ns) and >50 ns; (b) intensity image of PtL1Cl-labelled epidermis at progressively longer time-delays; (c) emission spectra of PtL1Cl-labelled (black) and unlabelled (red) epidermis; (d) higher-resolution intensity image; (e) lifetime distribution corresponding to (d); (f) kinetic traces from cellular and matrix regions on (e). | ||
 | ||
| Fig. 4 PtL1Cl-labelled liver tissue section. (a) Total intensity image; (b) time-gated image recorded at the time interval 50–2000 ns after 760 nm, ∼120 fs excitation; (c) lifetime map; (d) kinetic traces from cellular and matrix regions in (d), red line corresponds to the fit with the lifetime of the main component being 1.69 ± 0.02 μs. | ||
The lifetime map acquired at higher spatial resolution (Fig. 3d and 3e for skin; Fig. 4 for liver) establishes that the emission lifetime of the complex inside the nuclei (1.7 ± 0.05 μs) is significantly longer than in the cytoplasm and extracellular regions (1.3 ± 0.1 μs). The lifetime of PtL1Cl-labels in the cell nucleoli is the same for hepatocyte and for keratinocyte cells in fixed tissue sections, but considerably shorter than in the nucleoli of live cells.
In general, the distribution of the lifetime values for PtL1Cl in the tissues is much narrower than that in live cells. One possible reason is that the emissive Pt(II)-label is more uniformly exposed to oxygen in the uniformly permeable tissue sections in contrast to metabolically active and selectively permeable live cells. A significant increase in the emission lifetime of the Pt-label across the tissue sections was observed upon equilibrating within an atmosphere of 95% N2/5% CO2, reinforcing the potential of the Pt-probes as oxygen sensors. The fact that intracellular matrix is virtually unlabelled in liver, but significantly labeled in skin, highlights the sensitivity of the probes and the method to the microenvironment exemplified by the composition of the matrix protein (type-IV collagen and laminin in liver vs. predominantly cysteine-rich keratin in epidermis).34 Overall, TP-TREM imaging of skin and liver tissue sections labelled with PtLnCl offers (i) high contrast images virtually free of background emission; (ii) a clear discrimination of the keratinocyte/hepatocyte cell nuclei from the background; and (iii) a significant variation of emission lifetimes of the Pt-probe depending on the location.
Temporal discrimination of multiple labels
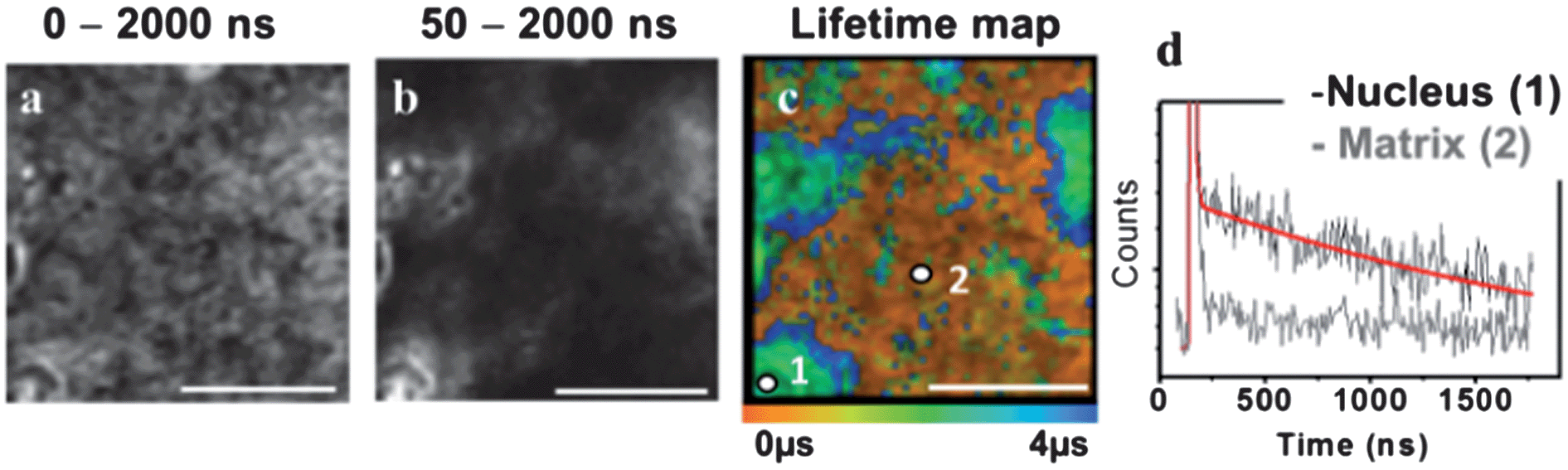
One of the key advantages of TP-TREM is its selectivity on the basis of the emission lifetime, applicable to simultaneous detection and discrimination of an array of labels which emit on significantly different time scales.35 We now further demonstrate the concept of temporal discrimination between a phosphorescent and a fluorescent probe under two-photon excitation. Live CHO-K1 cells were co-stained with PtL1Cl and with Hoechst-33342 – an intense, fluorescent nuclear stain with an emission lifetime of 2.2 ns. The Hoechst co-stain and PtL1Cl emit in the same region of the spectrum (∼500 nm), and both co-localise in the nucleus – thus providing the most challenging conditions where discrimination on the basis of the steady-state image or emission wavelength/colour is not possible. Discrimination on the basis of the lifetime is clear, as shown by lifetime maps at the short (0–50 ns) and long (50–2000 ns) time ranges, Fig. 5. The emission image accumulated over the short timescale corresponds almost exclusively to the fluorescence of Hoechst stain, and at long times, >50 ns, exclusively to emission of the Pt-label.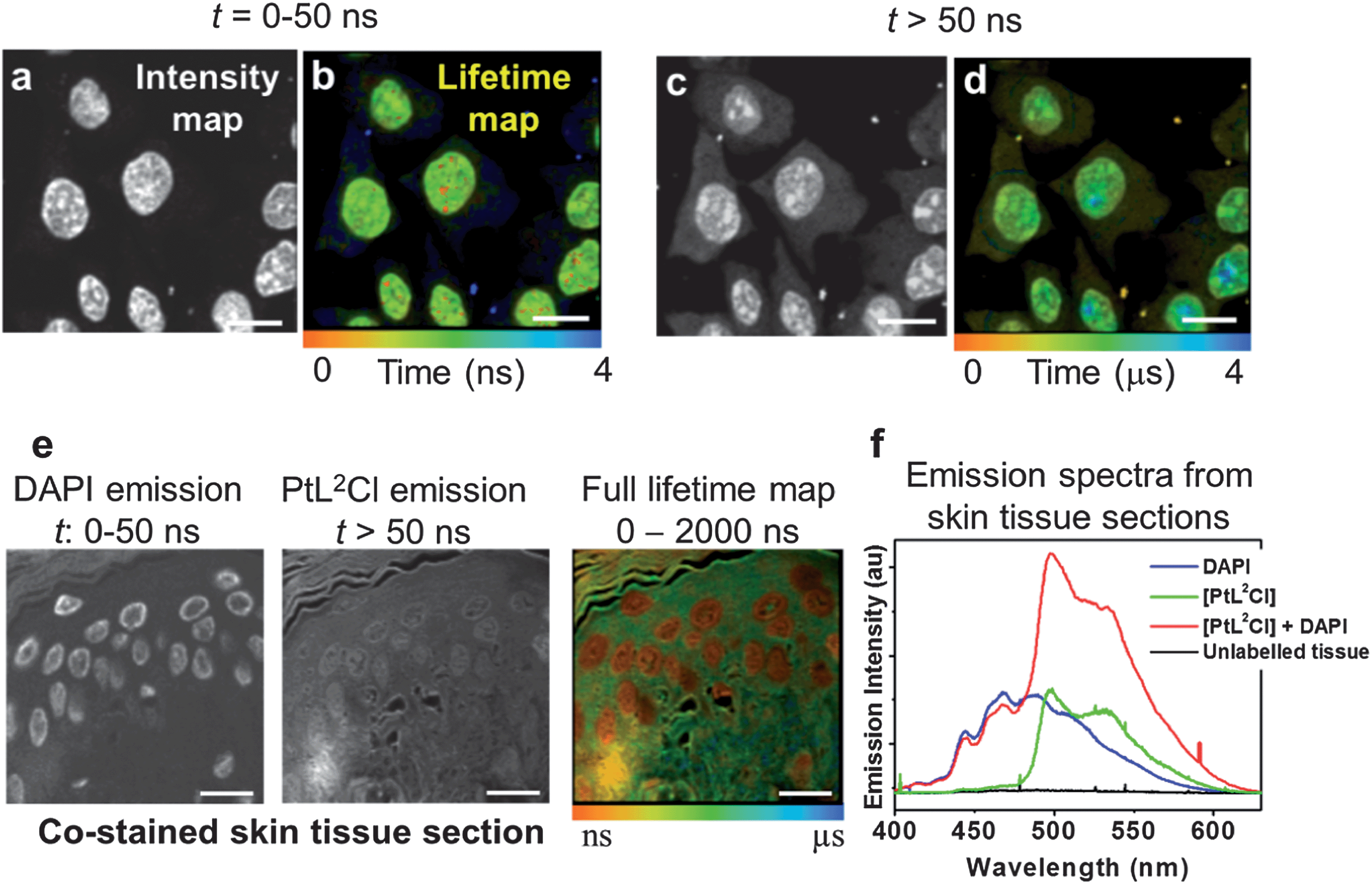 | ||
| Fig. 5 Discrimination between fluorescent and phosphorescent labels in multiply-stained cells and tissues. Top panel: two-photon TREM of live CHO-K1 cells co-labelled with PtL1Cl and Hoechst. Intensity images (a and c) and lifetime maps (b and d). Image at 0–50 ns is dominated by Hoechst emission. At >50 ns, the images (c and d) correspond to long-lived emission from PtL1Cl. Bottom panel: skin epidermis co-labelled with PtL2Cl and DAPI. (e) Time-gated intensity images demonstrate separation of short-lived emission of DAPI/autofluorescence from PtL2Cl. (f) Emission spectra from epidermis: unlabelled (black), labelled with DAPI (blue) or PtL2Cl (green), co-labelled with PtL2Cl and DAPI (red). | ||
The concept of temporal discrimination is further demonstrated on more complex biological structures, namely tissues. Skin tissue sections were dual-labelled with PtL2Cl and the blue-fluorescent organic nuclear stain DAPI (Fig. 5e and f) which co-localised in the cell nuclei. Lifetime mapping of the phosphorescent Pt-label was achieved by gating out the short-lived DAPI emission, yielding at longer time delay not only an autofluorescence-free but also a fluorescent-stain-free image. The complete discrimination between the fluorescent and the phosphorescent labels based on their different emission lifetimes allowed us to highlight different tissue sub-structures, and to create a Pt-label lifetime map (10 ns to 3 μs) without any background (Fig. 5).
Experimental details
PtLnCl (n = 1 and 2) complexes were synthesized and characterized as described previously.30 The new water-soluble derivative, PtL3Cl, was synthesised from ligand HL3 (ESI†). The two-photon absorption cross-section of PtL3Cl was determined using a comparative method.32 The cytotoxicity of PtL3Cl was assessed using MTT assay (Fig. S13 and S14†).Cell preparation and staining
CHO-K1 cells were cultured in a humidified 37 °C 5% CO2/95% air (v/v) environment in Ham cF12 medium (Sigma-Aldrich) supplemented with 10% (v/v) FCS, 100 units per ml penicillin, 100 μg ml−1 streptomycin, and 0.25 μg ml−1 amphotericin B. For microscopy, cells were passaged using 0.02% (v/v) EDTA and grown on 35 mm glass bottomed dishes (Mat Tek Corporation) until approximately 60% confluent. Cells were labelled with 100 μM PtL3Cl (in PBS) or 50 μM of PtL1/2Cl (in 1% DMSO/PBS) at 37 °C for 5 minutes and washed with PBS (3×). Cells were immersed in PBS during imaging.Tissue preparation and staining
Rat skin and liver tissues were processed by standard wax embedding, and histology microtome sections (10 μm) were taken. For microscopy, sections were de-waxed with xylene and serially re-hydrated (using ethanol at 100%, 90%, 70% and 50%, followed by PBS immersion for a minimum of 2 minutes each). Tissues were then labelled with (i) PtL1Cl by covering rehydrated sections with 200–400 μL of 100 μM PtL1Cl (2% DMSO in PBS) for 5 minutes at room temperature; (ii) 300 nM 4′,6-diamidino-2-phenylindole (DAPI) in PBS in conjunction with PtL1Cl for 5 minutes at room temperature, and (iii) using Hoechst 33342 (2′-[4-ethoxyphenyl]-5-[4-methyl-1-piperazinyl]-2,5′-bi-1H-benzimidazole trihydrochloride trihydrate) in PBS. For (i) and (ii), sections were washed with water and mounted directly by floating a cover slip over the tissue sample. For (iii), sections were mounted using a DPX mounted coverslip.Two-photon time-resolved emission imaging microscopy
Two-photon time-resolved microscopy and imaging of [PtLnCl] in solution, in viable mammalian cells, and in histological tissue sections were performed by using a mode-locked Ti-sapphire laser (FD900, Coherent UK) operating at 760 nm with pulse length ∼300 fs, pumped by a Vanadate V18 green continuous wave laser (Coherent UK). Due to the long excited state lifetime of the complexes (>3 μs), it was necessary to reduce the normally very high repetition rate of the Ti-sapphire laser from 76 MHz (13.1 ns between laser pulses) to that compatible with the emission lifetime. We note that in the data analysis, the value of the longer lifetime component may be underestimated due to the approx. 3-microsecond limit of the present set-up.The process of multiphoton absorption is critically dependent on the laser peak power, and so the energy per laser pulse was increased whilst reducing the repetition rate of the laser together with the average power at the sample. The Ti-sapphire laser was operated in a cavity dumped mode by extending the normal folded laser cavity of 1.8 m to more than 2.8 m (APE cavity dumper, Germany). The new lasing cavity contained a silica crystal which allowed variable laser repetition rates from 5.5 MHz to single shots when a high radio-frequency was applied. The laser was operated at 250 kHz with an average energy per pulse of 60 nJ (variable at the sample) compared to the pJ levels and >50 MHz repetition rate normally used for multiphoton excitation of biological samples. Time-resolved emission lifetime images were obtained using a modified two-photon-microscopy apparatus, constructed in the Central Laser Facility, which has an external x, y galvanometers scanning system (GSI Lumonics). Laser light was focused through a 60× water immersion objective with an NA of 1.2 on an inverted Nikon microscope (TE2000-U). The emitted light was collected without descanning, by-passing the scanning system, and passed through a bandpass filter (BG39, Comar). The scan was operated in the normal mode and line, frame and pixel clock signals were generated and synchronized with an external fast micro-channel plate photomultiplier tube (Hamamatsu R3809U) used as the detector. These were linked via a time-correlated single photon counting (TCSPC) PC module SPC830 (Becker and Hickl). The set-up provided instrument quantum efficiencies of more than 50% with single photon detection capabilities. The non-descan method also allowed increased signal detection over the normal one-photon method. Steady-state grey-scale multiphoton images (8 bit, up to 256 × 256 pixels) were produced by binning all decay photons as a single channel. Emission lifetime images were obtained by analysing the decay at individual pixels using a single or double exponential curve fitting following some modification to the standard Becker–Hickl SPCImage analysis software (B&H SPCImage 2.94) to allow analysis of the microsecond decay domain. A thresholding function within the analysis software ensured that non-correlating photons leading to background noise arriving at the detector were not included in the analysis. The lifetime image data are presented without further image processing.
Emission spectra under two-photon excitation were recorded by sending the emission signal from a specific pixel position through a spectrometer to a CCD detector setup (Andor iDUS) at another port of the microscope.
Conclusions
We bring together microsecond lifetime emission imaging with multiphoton excitation and sub-micron spatial resolution – two approaches that were previously incompatible – to generate a new powerful method of two-photon time-resolved emission imaging microscopy for life sciences. The new method expands the available imaging timescales 1000-fold whilst maintaining the essential spatial resolution, and allows complete background rejection. TP-TREM in conjunction with brightly emissive water-soluble microsecond probes allowed us to principally enhance the two key parameters in emission imaging, namely contrast and sensitivity to the local microenvironment in vivo, compared to existing methods.Here, we illustrate the power of TP-TREM through its application to a range of problems in biological imaging on the example of diverse live cells and histological tissues. Using long-lived, biocompatible platinum-based probes we demonstrate (i) high-contrast, autofluorescence-free multiphoton depth imaging of a range of live cell types and mammalian tissues; (ii) variation in the probe lifetime from nanoseconds to several microseconds depending on the localization, which highlights the sensing potential of the probes towards the local environment in vivo, and contrasts with the sub-nanosecond differences typically observed in the well-known technique of FLIM.
These results pave the way to background-free depth imaging of complex biological structures using the TP-TREM concept with an array of site-specific labels, which may differ in lifetime, localization, or emission wavelength (colour) and can be differentiated on the basis of lifetime differences in a single experiment, leading to multimodal sensing capability. For clinical and biological imaging, these may range from nanomolar sensing of bio-relevant analytes in vivo and fundamental FRET-type studies of biochemical processes, to the investigation of the evolution of tissue morphology and composition of matrix proteins associated with disease progression. Ultimately, this could provide autofluorescence-free visualization of surgical tumour treatments or monitoring of photodynamic therapy via oxygen detection.
The concept of TREM can be seen as “FLIM + PLIM” as it can be used on any timescale, from ultrafast fluorescence of organic molecules to slower emission of transition metal complexes or lanthanides/actinides, and combinations thereof. Exploiting the newly available plethora of microsecond metal-based labels, TP-TREM should be universally applicable to imaging any complex 3D-structure, which can also be subject to spatial and temporal evolution. The new approach of multiphoton-excitation/microsecond-detection brings transition metal probes to the forefront of lifetime imaging, and creates a transformative framework for microsecond multiphoton imaging across biomedical and material sciences.
Acknowledgements
We thank EPSRC, STFC, BBSRC, and the Universities of Sheffield and Durham for support and Prof A W Parker for discussions. We thank Becker & Hickl GmbH (Germany), and especially Dr Wolfgang Becker, for fruitful collaboration on the first implementation of the PLIM technique with scanning mode.Notes and references
- N. Johnsson and K. Johnsson, ACS Chem. Biol., 2007, 2, 31 CrossRef CAS PubMed.
- S. Berning, K. I. Willig, H. Steffens, P. Dibaj and S. W. Hell, Science, 2012, 335, 551 CrossRef CAS PubMed.
- (a) L. Bachmann, D. M. Zezell, A. D. Ribeiro, L. Gomes and A. S. Ito, Appl. Spectrosc. Rev., 2006, 41, 575 CrossRef CAS; (b) C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925 CrossRef CAS PubMed.
- T. J. Gould, M. S. Gunewardene, V. V. Verkhusha, J. A. Gosse and S. T. Hess, Nat. Methods, 2008, 5, 1027 CrossRef CAS PubMed.
- S. H. Shima, C. Xia, G. Zhong, H. P. Babcock and J. C. Vaughan, et al. , Proc. Nat. Acad. Sci. U. S. A., 2012, 109, 13978 CrossRef PubMed.
- (a) J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906 CrossRef CAS PubMed; (b) A. J. Lam, F. St. Pierre, Y. Y. Gong, J. D. Marshall, P. J. Cranfill, M. A. Baird, M. R. McKeown, J. Wiedenmann, M. W. Davidson, M. J. Schnitzer, R. Y. Tsien and M. Z. Lin, Nat. Methods, 2012, 9, 1005 CrossRef CAS PubMed; (c) Molecular Probes Handbook: A Guide to Fluorescent Probes and Labelling Technologies, ed. I. Johnson and M. T. Z. Spence, Invitrogen, 11th ed, 2010, ISBN: 978–0982927915 Search PubMed.
- G. Bunt and F. S. Wouters, Int. Rev. Cytol., 2004, 237, 205 CrossRef CAS.
- G. Marriott, R. M. Clegg, D. J. Arndt-Jovin and T. M. Jovin, Biophys. J., 1991, 60, 1374 CrossRef CAS.
- (a) J. W. Borst and A. J. W. G. Visser, Meas. Sci. Technol., 2012, 21, 102002 CrossRef; (b) P. A. Waghorn, M. W. Jones, M. B. M. Theobald, R. L. Arrowsmith, S. I. Pascu, S. W. Botchway, S. Faulkner and J. R. Dilworth, Chem. Sci., 2013, 4, 1430 RSC.
- K. Suhling, P. M. W. French and D. Phillips, Photochem. Photobiol. Sci., 2005, 4, 13 CAS.
- (a) R. M. Levenson, D. T. Lynch, H. Kobayashi, J. M. Backer and M. V. Backer, ILAR J., 2008, 49, 78 CrossRef CAS; (b) J. V. Frangioni, Mol. Imaging, 2009, 8, 303 Search PubMed.
- W. Zhong, P. Urayama and M. A. Mycek, J. Phys. Appl. Phys., 2003, 36, 1689 CrossRef CAS.
- J. Lecoq, A. Parpaleix, E. Roussakis, M. Ducros, Y. G. Houssen, S. A. Vinogradov and S. Charpak, Nat. Medods, 2011, 17, 893 CrossRef CAS PubMed.
- R. L. Jensen, J. Arnbjerg and P. R. Ogilby, J. Am. Chem. Soc., 2012, 134, 9820 CrossRef CAS PubMed.
- L. J. Charbonniere, N. Hildebrandt, R. F. Ziessel and H. G. Loehmannsroeben, J. Am. Chem. Soc., 2006, 128, 12800 CrossRef CAS PubMed.
- A. E. Soini, A. Kuusisto, N. J. Meltola, E. Soini and L. A. Seveus, Microsc. Res. Tech., 2003, 62, 396 CrossRef CAS PubMed.
- B. Song, C. D. B. Vandevyver, A. S. Chauvin and J. C. G. Bünzli, Org. Biomol. Chem., 2008, 6, 4125 CAS.
- (a) X. Q. Guo, F. N. Castellano, L. Li and J. R. Lakowicz, Biophys. Chem., 1998, 71, 51 CrossRef CAS; (b) H. C. Gerritsen, R. Sanders, A. Draaijer, C. Ince and Y. K. Levine, J. Fluoresc., 1997, 7, 11 CrossRef CAS.
- The concept is similar to that long-employed in time-resolved immunoassays using europium(III) ions, e.g. L. Seveus, M. Vaisala, S. Syrjanen, M. Sandberg, A. Kuusisto, R. Harju, J. Salo, I. Hemmila, H. Kojola and E. Soini, Cytometry, 1992, 13, 329 CrossRef CAS PubMed.
- R. R. De Haas, R. P. M. van Gijlswijk, E. B. van der Tol, J. Veuskens, H. E. van Gijssel, R. B. Tijdens, J. Bonnet, N. P. Verwoerd and H. J. Tanke, J. Histochem. Cytochem., 1997, 45, 1279 CrossRef CAS PubMed.
- (a) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 CrossRef CAS; (b) D.-L. Ma, V. P.-Y. Ma, D. S.-H. Chan, K.-H. Leung, H.-Z. He and C.-H. Luang, Coord. Chem. Rev., 2012, 256, 3087 CrossRef CAS PubMed.
- C. A. Puckett, R. J. Ernst and J. K. Barton, Dalton Trans., 2012, 39, 1159 RSC.
- V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186 RSC.
- Q. Zhao, C. H. Huang and F. Y. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC.
- (a) K. K. W. Lo, A. W. T. Choi and W. H. T. Law, Dalton Trans., 2012, 41, 6021 RSC; (b) C. Y.-S. Chung, S. P.-Y. Li, M.-W. Louie, K. K.-W. Lo and V. W. W. Yam, Chem. Sci., 2013, 4, 2453 RSC.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762 CrossRef CAS PubMed.
- P. Steunenberg, A. Ruggi, N. S. van den Berg, T. Buckle, J. Kuil, F. W. B. van Leeuwen and A. H. Velders, Inorg. Chem., 2012, 51, 2105 CrossRef CAS PubMed.
- T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148 CrossRef CAS PubMed.
- (a) S. S. Howard, A. Straub, N. G. Horton, D. Kbat and C. Xu, Nat. Photonics, 2013, 7, 33 CrossRef CAS PubMed; (b) N. W. Choi, S. S. Verbridge, R. M. Williams, J. Chen, J.-Y. Kim, R. Schmehl, C. E. Farnum, W. R. Zipfel, C. Fischbach and A. D. Stroock, Biomaterials, 2012, 33, 2710 CrossRef CAS PubMed.
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609 CrossRef CAS PubMed.
- S. W. Botchway, M. Charnley, J. W. Haycock, A. W. Parker, D. L. Rochester, J. A. Weinstein and J. A. G. Williams, Proc. Natl. Acad. Sci. U. S. A., 2008, 42, 16071 CrossRef PubMed.
- (a) A. A. Heikal and W. W. Webb, Proceedings of SPIE, 2002, 4812, 1 CrossRef CAS PubMed; (b) C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481 CrossRef CAS.
- F. M. Boisvert, S. van Koningsbruggen, J. Navascues and A. I. Lamond, Nat. Rev. Mol. Cell Biol., 2007, 8, 574 CrossRef CAS PubMed.
- This may reflect the affinity of this class of probes to thiols: S. J. Lippard, Acc. Chem. Res., 1978, 11, 211 CrossRef CAS.
- The concept of temporal discrimination of fluorescent and metal-based phosphorescent probes has previously been limited to one-photon excitation; e.g. L. Murphy, A. Congreve, L. O. Palsson and J. A. G. Williams, Chem. Commun., 2010, 46, 8743 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterisation and lifetime mapping of PtL3Cl in skin tissue and live HDF cells. PtL3Cl-labelled cell viability, determined by MTT assay. Steady state images of dual-stained tissue sections and emission spectra from [PtLnCl] labelled cells and tissue sections. A video of Z-scan projection of [PtLnCl]-labelled live CHO cells demonstrating 3D-imaging. See DOI: 10.1039/c3sc51875b |
| This journal is © The Royal Society of Chemistry 2014 |