New pyrido[3,4-b]pyrazine-based sensitizers for efficient and stable dye-sensitized solar cells†
Weijiang
Ying‡
a,
Jiabao
Yang‡
ab,
Mateusz
Wielopolski
c,
Thomas
Moehl
b,
Jacques-E.
Moser
c,
Pascal
Comte
b,
Jianli
Hua
*a,
Shaik M.
Zakeeruddin
*b,
He
Tian
a and
Michael
Grätzel
*b
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, Shanghai 200237, People's Republic of China. E-mail: jlhua@ecust.edu.cn; Fax: +86 21 64252758; Tel: +86 21 64250940
bLaboratory for Photonics and Interfaces, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: shaik.zakeer@epfl.ch; michael.graetzel@epfl.ch
cPhotochemical Dynamics Group, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland
First published on 20th September 2013
Abstract
A series of new pyrido[3,4-b]pyrazine-based organic sensitizers (PP-I and APP-I–IV) containing different donors and π-spacers have been synthesized and employed in dye-sensitized solar cells (DSSCs). The absorption spectra properties of dyes are analysed by density functional theory (DFT). The calculated results in combination with the experiments suggest that the absorption characteristics and excited state features will mainly be dominated by charge transfer transitions from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) and to higher LUMO orbitals. Furthermore, attaching the octyloxy groups significantly extends the π-conjugation of the donor in APP-IV, which raises the HOMO energy and facilitates its oxidation. As a consequence, APP-IV exhibits the lowest HOMO–LUMO energy gap among all dyes, which, in turn, corresponds well with the red shift of the absorption spectra. Transient photovoltage and photocurrent decay experiments as well as electrochemical impedance spectroscopy indicate that the electron lifetime and charge recombination resistance are increased due to the introduction of octyloxy chains on the donor unit, resulting in the high photovoltage based on APP-IV. It was found that APP-IV based DSSCs with liquid electrolyte display the highest power conversion efficiency (PCE) of 7.12%. Importantly, a PCE of 6.20% has been achieved for APP-IV based DSSCs with ionic-liquid electrolytes and retained 97% of the initial value after continuous light soaking for 1000 h at 60 °C. This renders these pyrido[3,4-b]pyrazine-based sensitizers quite promising candidates for highly efficient and stable DSSCs.
Introduction
After Grätzel and co-workers' first reports in 1991,1 for the recent past decades, dye-sensitized solar cells (DSSCs) have attracted significant attention as alternatives to conventional solar cells due to their facile and low-cost fabrication combined with high performance.2 Among all the components of a DSSC, the sensitizers play the key role in achieving high power conversion efficiencies (PCEs) and have, therefore, been intensively studied. To date, DSSCs based on ruthenium-complexes have shown certified PCE values of 11.9%,3 whereas zinc–porphyrin co-sensitized DSSCs hold a new PCE record of 12.3%.4Compared to the metal-complex sensitizers, alternative metal-free organic sensitizers5 have attracted increasing attention due to their high molar extinction coefficient, tunable absorption properties, relatively high efficiency and low cost. Commonly, such organic sensitizers are synthesized by designing a donor–π–acceptor (D–π–A) configuration,6 owing to their efficient intramolecular charge transfer (ICT) properties. Furthermore, it is well established that for efficient solar energy conversion it is necessary to utilize dyes that possess high charge carrier mobility, broad absorption spectra and long-term stability. Recently, a series of novel D–A–π–A organic dyes7 incorporating additional acceptors such as diketopyrrolopyrrole,8 bithiazole,9 isoindigo,10 benzothiadiazole,11 benzotriazole,12 and quinoxaline13 into the traditional D–π–A structure were reported and are receiving increasing attention. Inserting an auxiliary acceptor, which acts as an electron trap and increases the efficiency of charge separation facilitates electron transfer from the donor to the anchor. Other consequences of a second acceptor moiety are often broader spectral ranges and enhanced stabilities. Relatively high efficiencies of nearly 9% have already been reported on DSSCs based on this type of sensitizers.11a Such promising results drive the quest to find new and appropriate additional acceptors for such D–A–π–A type dye structures.
Pyrido[3,4-b]pyrazine (PP)14 exhibits a well-known electron-withdrawing effect due to its two symmetric unsaturated nitrogen atoms and the pyridine N-atoms. It has already been widely utilized and shows promising photovoltaic properties in polymer structures for NIR light-emitting diodes,15 field-effect transistors,16 two-photon absorption17 and bulk heterojunction solar cells.18 Thus, using PP units as components in organic sensitizers is a very attractive approach in terms of their diverse structural modifications and the intrinsic strong electron-deficient properties. To the best of our knowledge, the potential of the PP moiety as an auxiliary acceptor for DSSCs has not yet been explored.
The most popular methods of structural modification for fine-tuning the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of sensitizer dyes are by judiciously, varying the donor fragments and/or the conjugation of the π-spacer. Here, we report the facile synthesis and DSSC performance of a series of organic D–A–π–A type sensitizers based on the PP derivatives, containing triphenylamine/octyloxytriphenylamine as donors and cyanoacrylic acid as acceptors (PP-I and APP-I–IV, as shown in Fig. 1) are reported for the first time. This work aims to demonstrate how the variation of the donor structure and/or the heterocyclic linkers (thiophene, furan and benzene) impacts the overall photo conversion efficiency of these dyes.
 | ||
| Fig. 1 Molecular structures of the synthesized dyes. | ||
Alkoxyl chains or aryl groups were introduced into the triphenylamine donor or the PP unit to prevent aggregation of the dyes.8b The synthetic procedure to obtain the five PP-based dyes involves three major steps as shown in Scheme S1.†
Results and discussion
Synthesis
A two-step Suzuki coupling reaction with a dibromo-PP derivative resulted in the corresponding aldehyde precursors (4a–e) and Knoevenagel condensation with cyanoacetic acid, as shown in Scheme S1.† In the Suzuki coupling reaction, preferential oxidative coupling occurs at the more electron deficient 4-carbon position of pyridine derivates leading to the pyridal N-atoms proximal to the donor.19 It is important to highlight the regio-chemistry of 3a–c, where the pyridal N-atoms of each PP acceptor unit are orientated towards the triphenylamine donor moiety (proximal configuration). This conformation is observed from the corresponding 1H NMR (shown in Fig. S1†), as the δ data of the benzene attached to the PP-core shifted down-field (8.22–8.07 ppm), strongly affected by the nearby pyridal N-atoms. Such a phenomenon has also been observed in other small molecules containing a pyridine ring.20 All key intermediates and five organic PP-based sensitizers were fully characterized with 1H NMR, 13C NMR, and HRMS (see the ESI†).Ground-state features
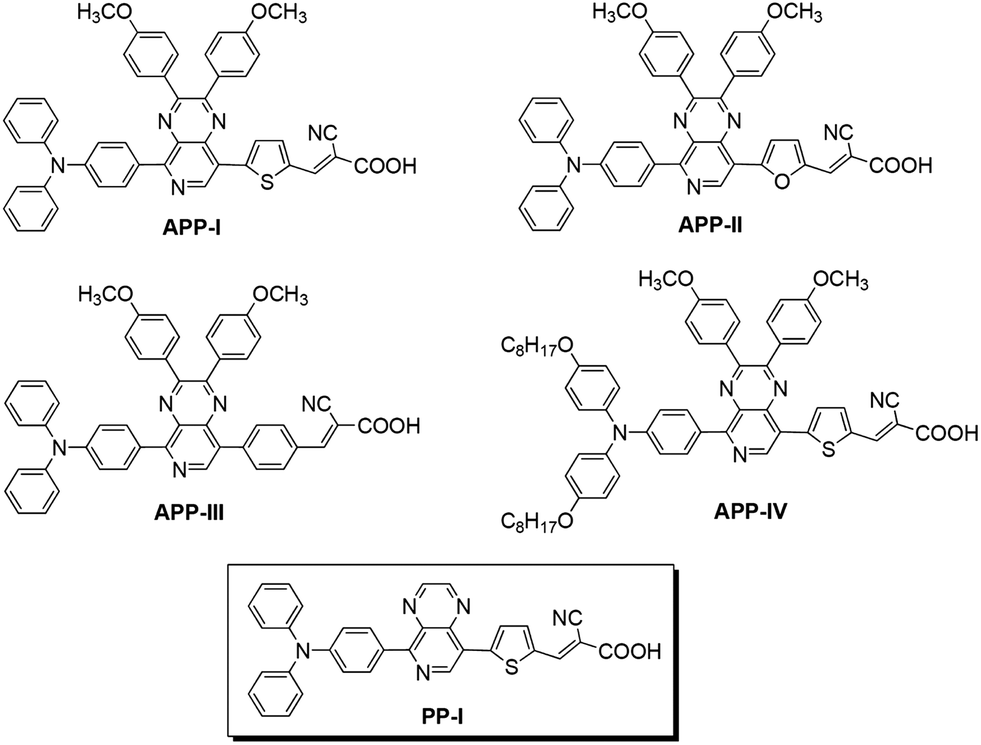
The absorption spectra of PP-based dyes in CH2Cl2 and as adsorbed onto 4 μm TiO2 films are shown in Fig. 2. Their absorptions, molar extinction coefficients, and HOMO–LUMO energy levels are summarized in Table 1. The UV-vis spectra exhibit two major prominent bands, one appearing at 360–400 and the other at 480–530 nm. The former can be ascribed to a localized aromatic π–π* transition from the triarylamine group to the PP moiety. The absorption at 480–530 nm may be attributed to the ICT transition from the triarylamine donor group to the cyanoacetic acceptor unit. The absorption maxima of PP-I and APP-I to IV appear at 500, 497, 506, 461, and 524 nm, respectively. A comparison of the absorption spectra of PP-I and APP-I, which differ solely by their PP cores, reveals very similar absorption properties, which suggests that changing the PP unit has hardly any effect on the shape of the spectra. | ||
| Fig. 2 (a) Absorption spectra of dyes in CH2Cl2; (b) absorption spectra of dyes on a 4 μm thin TiO2 films. | ||
| Dye | λ max a/nm (ε × 10−4 M cm) | λ max b/nm | HOMOc/V (vs. NHE) | E 0–0 d/eV | LUMOe/V (vs. NHE) |
|---|---|---|---|---|---|
| a Absorption maximum in CH2Cl2 solution (3 × 10−5 M). b Absorption maximum on TiO2 film. c HOMO potentials measured vs. Fc+/Fc were converted to those vs. the normal hydrogen electrode (NHE) by addition of +0.63 V. d E 0–0 was estimated from the intercept of the normalized absorption and emission spectra in each solvent used. e LUMO is calculated by subtracting E0–0 from the HOMO. | |||||
| PP-I | 423 (2.80) 500 (1.67) | 479 | 1.12 | 2.06 | −0.94 |
| APP-I | 396 (1.24) 497 (1.68) | 482 | 1.12 | 2.07 | −0.95 |
| APP-II | 431 (1.80) 506 (1.15) | 499 | 1.11 | 2.08 | −0.97 |
| APP-III | 396 (1.79) 461 (1.35) | 446 | 1.13 | 2.25 | −1.12 |
| APP-IV | 429 (2.20) 524 (2.33) | 516 | 0.85 | 1.95 | −1.10 |
On the contrary, varying the π-bridges (thiophene, furan and benzene) as is done for the three dyes APP-I to III leads to significant differences in the absorption features demonstrating that modulation of the electron density of D–A–π–A type dyes may be achieved through alterations of the π-bridges. Strikingly, modifying the triarylamine donor structure results in rather distinct spectral shifts. Upon changing the triphenylamine unit (APP-I) to a 4-octoxy-N-(4-octoxyphenyl)-phenylbenzeamine unit (APP-IV) the absorption edge shifts to the red by 27 nm. The reason behind this spectral shift is that the two octyloxy groups attached to the triphenylamine electron donor in APP-IV enhance the extent of electron delocalization over the entire donor moiety increasing the π-conjugation length, which, in turn, lowers the HOMO–LUMO gap, red-shifting the spectrum. Compared with the solution spectra, the maximum absorption peaks of PP-I and APP-I–IV on the 4 μm thin transparent TiO2 films shift to the blue by 21, 15, 13, 15, and 8 nm, respectively (Table 1). The blue shift in the absorption spectra of dyes adsorbed on the surface of the TiO2 film may be ascribed to the deprotonation of the carboxylic acid.21
Cyclic voltammetric data for the PP-I and APP-I to IV dyes is shown in Table 1 and Fig. S2.† The redox potentials (Eox) of PP-I and APP-I to IV corresponding to the HOMO level energy, are located at 1.12, 1.12, 1.11, 1.13, and 0.85 V, respectively, vs. a normal hydrogen electrode (NHE). These values are more positive than the redox potential of the I−/I3− redox couple (0.4 V vs. NHE) guaranteeing efficient dye regeneration. Interestingly, the HOMO level of APP-IV is less positive than those of the other dyes, which is ascribed to the stronger electron donor ability of the octyloxydiphenylamine unit. The zero–zero transition energies (E0–0) of the five dyes were determined to be 2.06, 2.07, 2.08, 2.25, and 1.95 V, respectively, which is estimated from the intercept of normalized absorption and emission spectra. The estimated excited-state potentials corresponding to the LUMO levels, calculated from EHOMO to E0–0, are −0.94, −0.95, −0.97, −1.12, and −1.10 V vs. NHE, respectively. The obtained LUMO values are much more negative than the Fermi level of TiO2 (−0.5 V vs. NHE), ensuring an efficient electron injection process from the excited state of the dyes into the TiO2 electrode.
Theoretical approach
Density functional theory (DFT) calculations were carried out to gain further insight into the molecular structures and the electronic distribution of the frontier molecular orbitals of the dyes. The structures of all dyes were optimized using the B3LYP22 functional with the 6-31G*23 basis set as implemented in the Gaussian 0924 suite of programs. All octyloxy groups have been replaced with methoxy groups to save calculation time. The resulting geometries are shown in Fig. 3. Apparently, the variation of the π-linker between the PP moiety and the cyanoacrylic acid anchoring moiety results in a variation of the distance between the triarylamine nitrogen and the carboxylic acid of the anchoring group. The geometry of the furan ring in APP-II obliges the molecular structure to bend with respect to the anchoring group, whereas the phenyl ring in APP-III results in a stretched configuration of the dye structure. The thiophene spacer in APP-I, APP-IV and PP-I forces the molecules to adopt an intermediate geometry somewhere in-between that of the bent conformation of APP-II and the stretched confirmation of APP-III. Apart from a change in the distance between the electron donating triarylamine unit and the electron accepting anchoring group, such geometrical variations will have an impact on the adsorption geometries on TiO2. For instance, closer contacts to the TiO2 surface as imposed by the structures of APP-I, APP-II, APP-IV and PP-I may render back-electron transfer more likely than in the stretched APP-III dye. The phenyl ring in APP-III induces a significant increase of the dihedral angle between the PP-unit and the π-spacer to a value of 42.8°, which is nearly 20° more than that found with APP-I (23.5°). Consequently, the overlap of the p-orbitals of the spacer and the PP-moiety is reduced in APP-III as compared to the other dyes.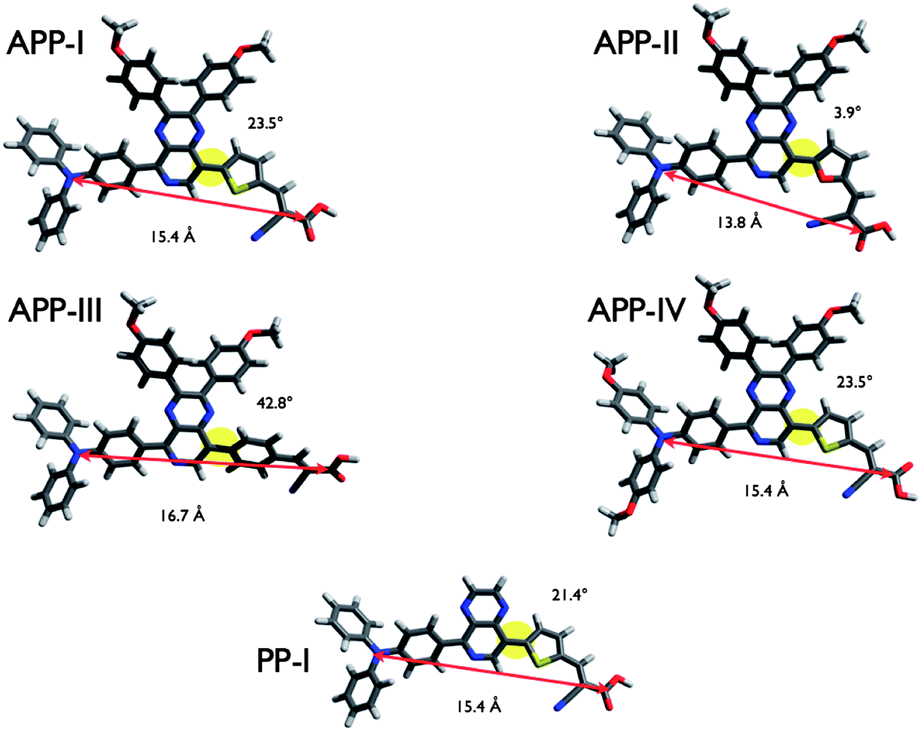 | ||
| Fig. 3 B3LYP/6-31G* molecular geometries of all PP sensitizers as resulted from DFT calculations. The arrows indicate the varying center-to-center distances from the triarylamine nitrogen atom to the carboxylic acid carbon of the anchoring group. The dihedral angles between the π-spacer and the PP cores are indicated on top of the structures. | ||
The influences of the variations of the chemical structure are further apparent when considering the electronic properties of the dyes, i.e. the frontier orbitals and their energies. The four highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) with their corresponding energies and the HOMO–LUMO gaps are depicted in Fig. 4. All dyes exhibit a typical donor–acceptor-type architecture with the HOMO concentrated on the electron donating triarylamine moiety and the LUMO on the electron accepting cyanoacrylic acid anchor.
 | ||
| Fig. 4 Representations of the electron densities of the four highest occupied molecular orbitals (HOMO to HOMO−3) and the four lowest unoccupied molecular orbitals (LUMO to LUMO+3) as resulted from B3LYP/6-31G* geometry optimization of all PP sensitizers. The corresponding HOMO–LUMO energy gaps are given in red. | ||
Detailed considerations on the orbital energies and their electron density distribution (see ESI†) come to the conclusion that the absorption characteristics and excited state features will mainly be dominated by charge transfer transitions from the HOMO to LUMO and to higher LUMO orbitals. This is in conjunction with former results from TD-DFT calculations on similar quinoxaline-based D–A–π–A sensitizers.7,13b Furthermore, attaching the octyloxy groups significantly extends the π-conjugation of the donor in APP-IV, which raises the HOMO energy and facilitates its oxidation. As a consequence, APP-IV exhibits the lowest HOMO–LUMO energy gap (2.08 eV) among all dyes, which, in turn, corresponds well with the red shift of the absorption spectra (Fig. 2).
Photovoltaic performance
The DSSC performances of all the dyes were tested under AM 1.5G irradiation (1 sun, 100 mW cm−2). To prevent inflated photocurrents arising from stray light, a black metal mask surrounded the active area.25 The photocurrent density voltage curves (J–V) and the incident photon to current conversion efficiency (IPCE) spectra of the DSSCs based on the PP dyes are shown in Fig. 5. The detailed parameters of short-circuit current density (Jsc), open-circuit voltage (Voc), fill factor (ff), and power conversion efficiency (PCE) are collected in Table 2. Hereby, the influence of altering the PP core, π-bridges and donors on the DSSC characteristics is put into focus. When compared with PP-I, APP-I shows both higher Jsc and Voc due to higher electron injection efficiency (87.4 vs. 52.6, shown in Table S1†) and less aggregation due to the insertion of the 4-methoxyphenyl groups on the PP unit. This phenomenon can also be understood by the fact that the APP-I based cells show a much higher IPCE value than the PP-I based cells in the range of 400–600 nm. | ||
| Fig. 5 (a) J–V curves for DSSCs based on the dyes under illumination of AM 1.5G simulated sunlight (100 mW cm−2). (b) IPCE spectra of the same DSSCs. | ||
| Dye | J sc (mA cm−2) | V oc (mV) | ff | η (%) |
|---|---|---|---|---|
| a Electrolyte: 0.03 M I2, 0.05 M LiI, 0.5 M TBP, 0.1 M GNCS, 1.0 M DMII in acetonitrile and valeronitrile (v/v, 85/15). | ||||
| PP-I | 7.10 | 570 | 0.76 | 3.11 |
| APP-I | 12.11 | 671 | 0.76 | 6.14 |
| APP-II | 8.37 | 631 | 0.73 | 3.93 |
| APP-III | 8.20 | 694 | 0.76 | 4.35 |
| APP-IV | 13.56 | 691 | 0.76 | 7.12 |
Furthermore, the effect of varying the π-bridges is highlighted in terms of the changes in Voc. Scrutinizing the dyes with different linkers (APP-I with thiophene, APP-II with furan, and APP-III with benzene), it is apparent that the Voc of APP-III is higher than those of the APP-I and APP-II based devices. This may be caused by the introduction of a benzene group, which leads to a reduction of electron recombination and improves the Voc due to the relatively large dihedral angle between the PP unit and the benzene-bridge (see Theoretical approach). Such a phenomenon has been also observed in other dyes.10 It should be noted that the PCE of the APP-III based cell was not the highest due to its relatively narrow absorption range and the lower Jsc. In general, the Jsc is related to the molar extinction coefficients and the absorption spectra of the dyes. Accordingly, a higher molar extinction coefficient yields a higher short-circuit current. The photocurrent of the APP-IV cell with 4-(bis(4-octoxylphenyl)amino)phenyl as electron donor was higher than that for the APP-I which contains the triphenylamine as donor. This may be attributed to the higher molar extinction coefficient and the broader absorption of APP-IV, which yields better light harvesting and higher Jsc. In addition, the much more negative LUMO energy level of APP-IV leads to a better electron injection efficiency (82.9 vs. 87.4, shown in Table S1†) from the excited state of the sensitizer to the conduction band (CB) of TiO2. Moreover, the APP-IV based cells show higher Voc than the APP-I cells. This could be explained by the fact that the two octyloxy chains on the 4-(bis(4-octyloxphenyl)amino)phenyl of APP-IV help to form a blocking layer which limits the access of I3− ions to the TiO2 electrode surface, hence increasing the electron lifetime and the Voc. The Jsc values of all five of the devises are consistent with the resulting IPCEs.
Laser studies
Nanosecond laser flash photolysis measurements were carried out to observe the impact of the structural modifications on the reaction rates of both the charge recombination between the injected electron ecb− and oxidized radical cation of the sensitizer S+ (eqn (2)) and the regeneration of the dye by the redox electrolyte (eqn (3)).| S*|TiO2 → S+|TiO2 + ecb− (TiO2) | (1) |
| S+|TiO2 + ecb− (TiO2) → S|TiO2 | (2) |
| S+|TiO2 + D → S|TiO2 + D+ | (3) |
| ecb− (TiO2) + D+ → D | (4) |
Both reactions follow the ultrafast charge injection from the excited state of the dye S* into the TiO2 conduction band (eqn (1)) and compete kinetically.26 For these studies, the various sensitizers were adsorbed on 4 μm thick transparent TiO2 films and excited at a wavelength of 530 nm using laser pulses with a duration of 7 ns. In order to scrutinize both processes, i.e. the back-electron transfer between the injected electrons and the oxidized state of the dye and the reduction of the latter by the donor D contained in the redox electrolyte (eqn (2) and (3)), the studies were performed in the presence and the absence of the redox electrolyte solution.
All dyes exhibit comparable spectral features in their excited states. A representative transient absorption spectrum of PP-I in the absence of the electrolyte is shown in Fig. 6. The broad transient absorption feature developing at wavelengths above 620 nm is characteristic of the triarylamine cation of the oxidized dye molecule S+.27 Hence, following the decay kinetics of the transient absorption signal measured at 680 nm allowed for the analysis of the charge recombination and dye regeneration rate constants, and for the determination of the half-reaction time of both deactivation processes – t1/2,rec in the absence of the redox electrolyte and t1/2,reg in the presence of the redox electrolyte solution.
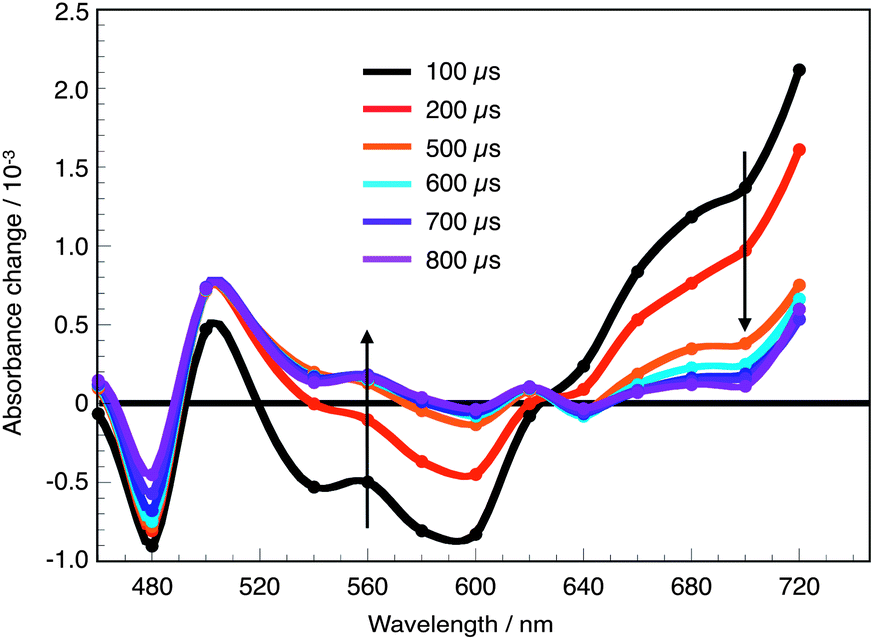 | ||
| Fig. 6 Transient absorption spectra measured upon nanosecond laser excitation (λexc = 530 nm) of PP-I adsorbed on 4 μm mesoporous TiO2 films in the absence of redox electrolyte at various delay times between 100 μs and 1 ms. All dyes show comparable spectral signatures. | ||
Upon photoexcitation of the sensitizer, an electron is readily injected into the TiO2 film (eqn (1)). In a pure solvent and in the absence of a donor, the injected electrons have no other possibility than recombining with the oxidized dye cation S+.
Fig. 7a displays the time-profiles of the transient absorption signal measured at 680 nm in the absence of a redox electrolyte for all investigated compounds. The corresponding half-reaction times are listed in Table 3. The results reveal that upon modification of the PP-core, the charge recombination time constants do not change significantly. From current electron transfer dynamics theories, it is expected that the charge recombination rate decreases exponentially when the distance separating the oxide surface from the triarylamine moiety of the dye radical cation is increased, provided the energetics of the process and the associated nuclear reorganization energy are comparable. APP-III dye is characterized by a larger distance separating the cyanoacrylate anchor from the amine group (16.7 Å instead of 13.8 Å for APP-II and 15.4 Å for APP-I, APP-IV, and PP-I). In this case the time constant for recombination t1/2,rec is indeed observed to be larger by a factor of ∼1.5 compared to the other compounds. This difference, however, is rather small compared to the factor of 5–30 that would be expected for through-space electron transfer with an ET distance elongated by 1.3–2.9 Å and a distance decay constant of 1.2 Å−1. The charge recombination kinetics appears then to be essentially controlled by the conformations of dye molecules adsorbed on the TiO2 surface, which all result in rather similar electron transfer distances.
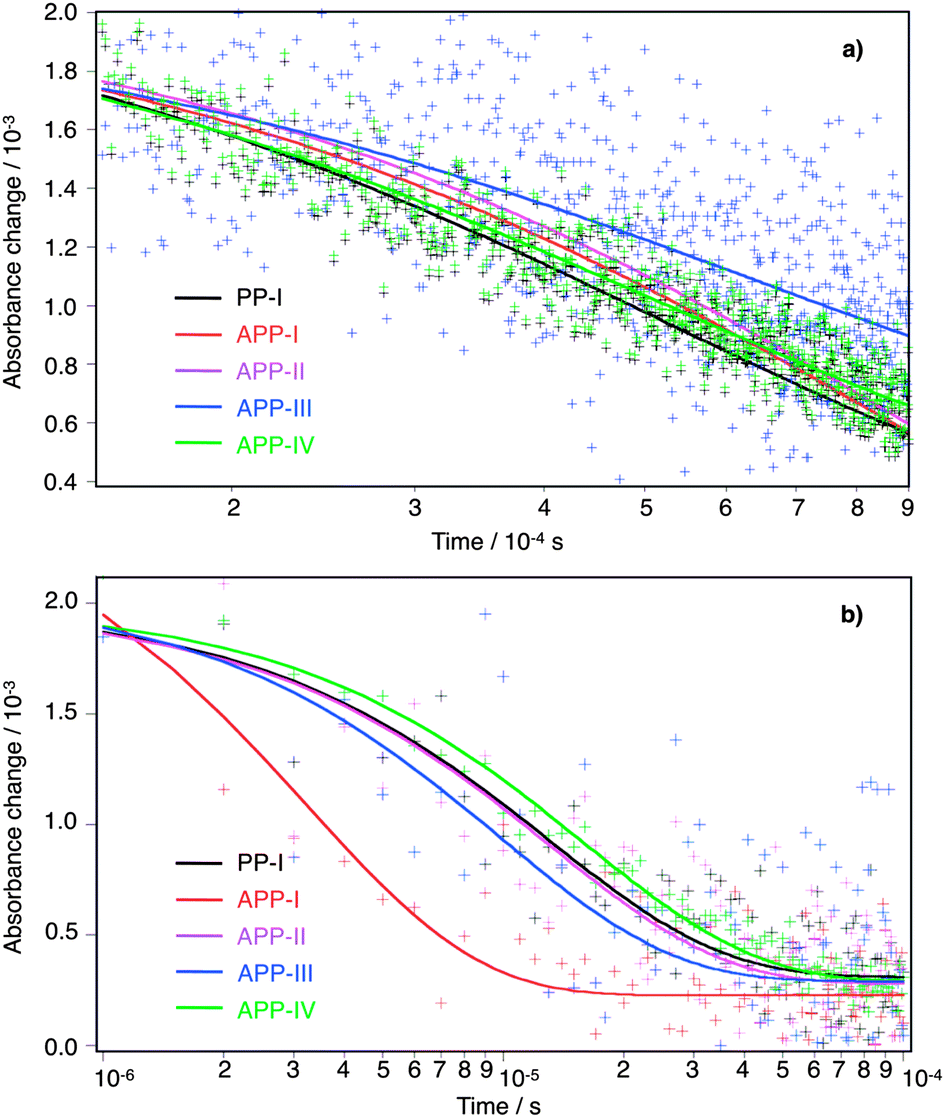 | ||
| Fig. 7 Transient absorbance decays of the oxidized state after nanosecond flash photolysis (λexc = 530 nm) of the PP sensitizers adsorbed on 4 μm nanocrystaline TiO2 films. The kinetics was monitored at 680 nm, (a) in the absence (no iodide electrolyte added) and (b) in the presence of the redox electrolyte Z960 under similar conditions. | ||
| PP-I | APP-I | APP-II | APP-III | APP-IV | |
|---|---|---|---|---|---|
| t 1/2,rec | 405 μs | 450 μs | 490 μs | 600 μs | 410 μs |
| t 1/2,reg | 9.8 μs | 3.1 μs | 9.5 μs | 7.5 μs | 10.6 μs |
Considering the dynamics in the presence of the redox electrolyte solution (Fig. 7b), a much faster decay of the dyes' oxidized states was observed, showing that the reduction of dye cations by the redox electrolyte (dye regeneration, eqn (3)) is competing efficiently with the charge recombination process. The dye regeneration of the PP-dye cation occurs within 3–11 μs, indicating that it is ca. 2 orders of magnitude faster than charge recombination.
In summary, the laser studies show that the differences observed in the photovoltaic performance of the DSSCs do not depend on the charge recombination in the absence of redox electrolyte or dye regeneration kinetics in the presence of the redox electrolyte. Characteristics such as ground state absorption properties or electronic features of the dyes play a major role in the photovoltaic performance of the dyes. However, charge recombination and dye regeneration are certainly governed by the chemical structures of the dyes – especially with respect to its impact on the adsorption geometry on the TiO2 surface or shielding of the positive charge of the triarylamine cation.
Transient photovoltage and photocurrent as well as electrochemical impedance (EIS) spectroscopy were performed to develop a more in-depth understanding about the influence of different dyes on the recombination (eqn (4)), the transport properties and the conduction band edge position of the TiO2 film. The dark current characteristics of the different devices and the parameters such as the transport and the recombination resistance as well as the chemical capacitance, were extracted by fitting the EIS data with the transmission line model, are presented in the ESI (Fig. S3 and S4†). In the low forward bias region, dominated by the FTO–electrolyte interface, the dark current is smallest for APP-IV and APP-II, followed by APP-I, APP-III and PP-I. At higher forward bias, i.e., around the maximum power point, the smallest dark current is measured for the APP-III device and the highest for PP-I device, while APP-I, APP-II and APP-IV all show similar mid-range values.
The Jsc and Voc values of all the PP devices are tabulated in Table 2. The divergence in the Jsc values of various devices can be explained due to the differences in the spectral response of the PP dyes. The device's open-circuit potential (Voc) is influenced by the conduction band (Ecb) position and the recombination of electrons at the TiO2–electrolyte interface. The highest value of Voc obtained is for the devices fabricated with APP-III and APP-IV and the lowest for the PP-I device. Plotting the device capacitances against voltage indicates (Fig. 8) that the Ecb of the PP-I is approximately 45 mV lower than that of the APP-IV device. The downward shift of the Ecb for the PP-I device is partially responsible for the lower Voc (120 mV) measured in comparison with the APP-IV device.
 | ||
| Fig. 8 Applied voltage (corrected for IR drop) plotted versus the chemical capacitance determined by impedance spectroscopy. | ||
The electron lifetime (τn = CchemRct) versus the chemical capacitance is plotted for all the devices in order to compare the differences in the electron recombination processes. The chemical capacitance can be used as an indirect measure of the Fermi level, free of IR drop effects (under the assumption of a similar depth of the distribution of the trap states in the DOS) and is, therefore, the preferable representation form for analysing the changes of the electron lifetime.
At higher applied forward bias the electron lifetime is greatest for the APP-III device, followed in descending order by APP-IV, APP-I, APP-II and PP-I (Fig. 9a) devices. The dyes PP-I, APP-I and APP-IV all contain thiophene as a π-bridge in which PP-I is structurally the smallest molecule. APP-I has two additional 4-methoxyphenyl groups on the PP unit and APP-IV (similar base structure to APP-I) contains two additional octyloxy chains on the triarylamine donor moiety. The bulkier the size of the dye, the less of it should adsorb on the TiO2 films. The dye concentration measured on the surface of the TiO2 film for PP-I and APP-I dyes is similar, but it is lower for the APP-IV dye, in agreement with this reasoning. Insertion of 4-methoxyphenyl groups to the structure of PP-I to form APP-I helped in retarding the electron recombination process. The transformation of the APP-I dye structure to APP-IV through introduction of octyloxy chains on the donor unit helped further to retard the recombination process. The lowered recombination follows from the reduced access of triiodide present in the redox electrolyte to the TiO2 surface. The electron lifetimes obtained from the transient photovoltage and photocurrent measurements (Fig. 9b) matched well with the electron lifetime trends observed with the EIS measurements.
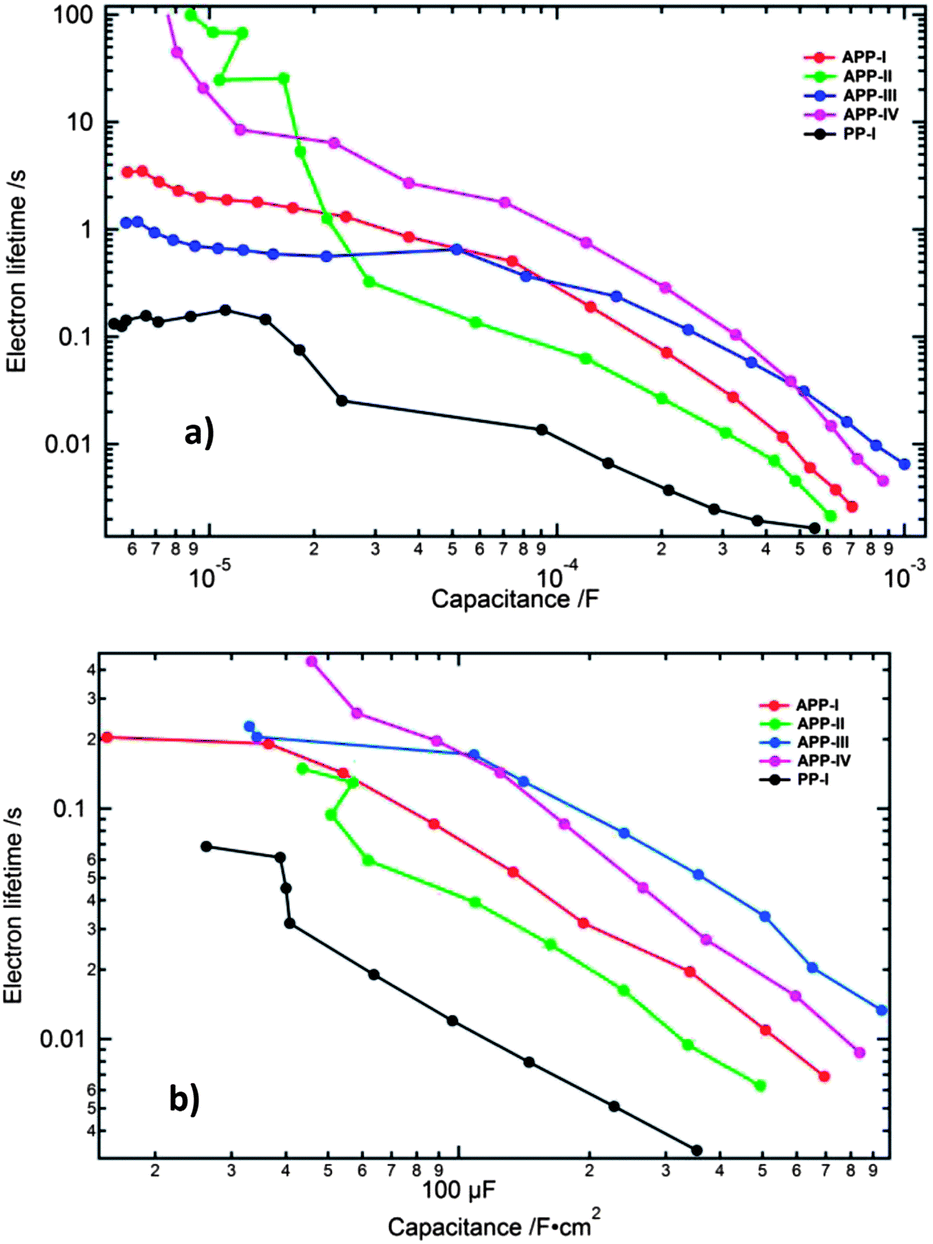 | ||
| Fig. 9 Electron lifetime plotted versus the chemical capacitance (a) from EIS measurements and (b) from the transient photovoltage and photocurrent decay measurements. | ||
Stability measurements
Long-term stability is a vital parameter for sustained cell operation. Substitution of the liquid electrolyte by a solvent-free ionic liquid allows the determination of the stability of the photovoltaic performance over an extended period of time. For the evaluations, APP-I and APP-IV were utilized as sensitizers in ionic liquid electrolyte DSSCs exploring 1,3-dimethylimidazoliumiodide/1-ethyl-3 methylimidazoliumiodide/1-ethyl-3-methyl-imidazolium tetracyanoborate/iodine/N-butylbenzoimidazole/guanidinium thiocyanate (molar ratio 12/12/16/1.67/3.33/0.67) as redox electrolyte. The photovoltaic parameters of devices using ionic liquid electrolyte measured under standard AM 1.5G full sunlight conditions are summarized in Table 4. These APP-I and APP-IV based DSSCs obtained PCE values of 4.96% and 6.20%, respectively. These devices were subjected to long-term stability tests under the irradiance of AM 1.5G full sun visible light soaking at 60 °C. The detailed photovoltaic parameters of the devices during the aging process are plotted in Fig. 10. The PCE of APP-I and APP-IV dyes containing devices remained at 100% and 97% of the initial value under both the thermal and the light-soaking stress for over 1000 h. This result indicates the underlying potential of PP-based dyes as promising candidates for highly efficient and stable DSSC devices.| Dye | J sc (mA cm−2) | V oc (mV) | ff | η (%) |
|---|---|---|---|---|
| a Electrolyte: 1,3-dimethylimidazoliumiodide/1-ethyl-3-methylimidazoliumiodide/1-ethyl-3-methylimidazoliumtetracyanoborate/iodine/N-butylbenzoimidazole/guanidiniumthiocyanate (molar ratio 12/12/16/1.67/3.33/0.67). | ||||
| APP-I | 9.61 | 647 | 0.78 | 4.96 |
| APP-IV | 12.28 | 648 | 0.76 | 6.20 |
 | ||
| Fig. 10 Stability test illustrating photovoltaic parameter (Jsc, Voc, ff, and η) variations with aging time for the devices based on (a) APP-I-, (b) APP-IV-sensitized TiO2 films with a solvent-free ionic-liquid electrolyte during 1 sun visible light irradiation at 60 °C. | ||
Conclusions
In summary, we have synthesized a series of new pyrido[3,4-b]pyrazine-based metal-free organic dyes and studied the impact of the different substitutions on the PP-core molecule, as well as the utilization of different π-spacers, and various donor moieties on the performance of DSSCs. The photovoltaic experiments show that the methoxyphenyl-substituted PP unit can improve the cells' Jsc and Voc, while the use of phenyl π-spacers rather than thiophene or furan moieties results mainly in an increased Voc. Moreover, the introduction of octyloxydiphenyl-amine groups as electron donors resulted in improved Jsc and Voc, contributing to the highest PCE (7.12%) for APP-IV based DSSC. EIS and transient photovoltage and photocurrent decay measurements demonstrated that the variations in Voc originated mainly from changes in the recombination behavior. A PCE of 6.20% has been achieved for the APP-IV sensitized DSSC in conjunction with an ionic-liquid electrolyte-based DSSC. This parameter remained at 97% of the initial value following continuous simultaneous thermal and light soaking stress for 1000 h. This work emphasizes the potential of using metal-free organic PP-based dyes for the development of efficient and stable DSSCs. Fine-tuning of the molecular structures provides experimental freedom to attain higher efficiencies and stable DSSC device performance.Acknowledgements
This work was supported by NSFC/China (2116110444, 21172073 and 91233207), the National Basic Research 973 Program (2013CB733700), and the Fundamental Research Funds for the Central Universities (WJ0913001). MG would like to thank the Swiss National Science Foundation and ECR advanced Grant Agreement no. 247404 under the CE-Mesolight project funded by the European community's 7th FWP for the financial support. J.-E. M. thanks NCCR MUST, a research instrument of the Swiss National Science Foundation for generous support. Yang would like to thank the funding of the visiting program which is supported by China Scholarship Council (CSC). We thank Dr Carole Grätzel at the EPFL for fruitful discussion.Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef
.
-
(a) M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed
-
(a) F. F. Gao, Y. Wang, D. Shi, J. Zhang, M. K. Wang, X. Y. Jing, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 10720–10728 CrossRef CAS PubMed
- A. Yella, H. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. G. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed
-
(a) A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed
-
(a) Y. P. Hong, J.-Y. Liao, D. R. Cao, X. F. Zang, D.-B. Kuang, L. Y. Wang, H. Meier and C.-Y. Su, J. Org. Chem., 2011, 76, 8015–8021 CrossRef CAS PubMed
- Y. Z. Wu and W. H. Zhu, Chem. Soc. Rev., 2013, 42, 2039–2058 RSC
-
(a) S. Y. Qu, W. J. Wu, J. L. Hua, C. Kong, Y. T. Long and H. Tian, J. Phys. Chem. C, 2010, 114, 1343–1349 CrossRef CAS
-
(a) J. X. He, W. J. Wu, J. L. Hua, Y. H. Jiang, S. Y. Qu, J. Li, Y. T. Long and H. Tian, J. Mater. Chem., 2011, 21, 6054–6062 RSC
- W. J. Ying, F. L. Guo, J. Li, Q. Zhang, W. J. Wu, H. Tian and J. L. Hua, ACS Appl. Mater. Interfaces, 2012, 4, 4215–4224 CAS
-
(a) W. H. Zhu, Y. Z. Wu, S. T. Wang, W. Q. Li, X. Li, J. Chen, Z. S. Wang and H. Tian, Adv. Funct. Mater., 2011, 21, 756–763 CrossRef CAS
-
(a) Y. Cui, Y. Z. Wu, X. F. Lu, X. Zhang, G. Zhou, F. B. Miapeh, W. H. Zhu and Z. S. Wang, Chem. Mater., 2011, 23, 4394–4401 CrossRef CAS
-
(a) D. W. Chang, H. J. Lee, J. H. Kim, S. Y. Park, S.-M. Park, L. M. Dai and J.-B. Baek, Org. Lett., 2011, 13, 3880–3883 CrossRef CAS PubMed
-
(a) B.-L. Lee and T. Yamamoto, Macromolecules, 1999, 32, 1375–1382 CrossRef CAS
- B. C. Thompson, L. G. Madrigal, M. R. Pinto, T.-S. Kang, K. S. Schanze and J. R. Reynolds, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1417–1431 CrossRef CAS
-
(a) P.-T. Wu, F. S. Kim, R. D. Champion and S. A. Jenekhe, Macromolecules, 2008, 41, 7021–7028 CrossRef CAS
-
(a) X. B. Huang, Q. Q. Shi, W.-Q. Chen, C. L. Zhu, W. Y. Zhou, Z. Zhao, X.-M. Duan and X. W. Zhan, Macromolecules, 2010, 43, 9620–9626 CrossRef CAS
-
(a) N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletete, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed
- S. T. Handy, T. Wilson and A. Muth, J. Org. Chem., 2007, 72, 8496–8500 CrossRef CAS PubMed
- Y. Sun, G. Welch, W. Leong, C. Takacs, G. Bazan and A. Hegger, Nat. Mater., 2012, 11, 44–48 CrossRef CAS PubMed
- Z. S. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2007, 111, 7224–7230 CAS
- P. J. Stephens, F. J. Devlin, C. F. Chablowski and M. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS
-
M. J. Frisch, et al., Gaussian, Inc., Wallingford CT, 2009
- S. Ito, M. K. Nazeeruddin, P. Liska, P. Comte, R. Charvet, P. Pechy, M. Jirousek, A. Kay, S. M. Zakeeruddin and M. Grätzel, Prog. Photovolt. Res. Appl., 2006, 14, 589–601 CrossRef CAS
- M. Sonntag, K. Kreger, D. Hanft, P. Strohriegl, S. Setayesh and D. Leeuw, Chem. Mater., 2005, 17, 3031–3039 CrossRef CAS
-
(a) M. Oyama, T. Higuchi and S. Okazaki, Electrochem. Solid-State Lett., 2002, 5, E1–E3 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sc51844b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |