
High yield of renewable hexanes by direct hydrolysis–hydrodeoxygenation of cellulose in an aqueous phase catalytic system†
Abstract
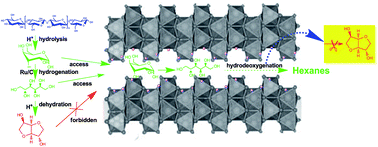
In aqueous phosphoric acid, cellulose was efficiently converted into hexanes using a Ru/C catalyst combined with layered compounds or silica–alumina materials. In this process, the direct production of hexanes from cellulose can be improved by suppressing the formation of isosorbide, which makes it difficult to yield hexanes by further hydrodeoxygenation. As the co-catalyst, layered compounds showed a significant inhibition effect on the formation of isosorbide from sorbitol due to the steric restrictions of sorbitol dehydration within the interlayers of layered compounds. Typically, layered LiNbMoO6 played a great role in promoting the production of hexanes directly from cellulose and a promising yield (72% carbon mol) of hexanes was obtained. In addition, the protonic acid, H3PO4, offered efficient catalysis for the hydrolysis of cellulose and the dehydration of the sorbitol hydroxyl moiety.
 Please wait while we load your content...
Please wait while we load your content...

