
Surface core–shell magnetic polymer modified graphene oxide-based material for 2,4,6-trichlorophenol removal
Abstract
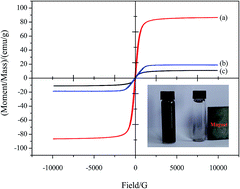
A novel well-designed graphene oxide-based magnetic polymer (GO-Fe3O4@P) has been successfully synthesized via distillation–precipitation polymerization, ring-opening and amidation reactions. The as-prepared GO-Fe3O4@P was further characterized by transmission electron microscopy (TEM), vibrating sample magnetometer (VSM), BET analysis, zeta potential analysis, X-ray photoelectron spectroscopy (XPS), and Fourier transform infrared spectroscopy (FTIR) and the characterization results revealed that core–shell structural Fe3O4@P microspheres were covalently bonded onto the GO sheet. The adsorption characteristics of the GO-Fe3O4@P intended for removal of 2,4,6-trichlorophenol (2,4,6-TCP) were investigated. Batch adsorption studies were carried out to optimize adsorption conditions. The effect of solution pH, adsorption isotherm, kinetics, and thermodynamics was deeply investigated. The results indicated that the adsorption property of GO-Fe3O4@P was highly pH dependent and the adsorption mechanism referred to hydrogen bond and π–π stacking interactions. The adsorption data for 2,4,6-TCP onto GO-Fe3O4@P was well fitted to a Langmuir isotherm. The maximum adsorption capacity (qm) of GO-Fe3O4@P to 2,4,6-TCP was found to be 232.6 mg g−1. Kinetic results showed that the adsorption reached equilibrium within 4 min, 10 min, and 60 min for initial 2,4,6-TCP concentrations of 10 mg L−1, 100 mg L−1, and 500 mg L−1, respectively. The data of adsorption kinetics obeyed the pseudo-second-order rate model well. Thermodynamic parameters such as ΔHθ, ΔSθ, and ΔGθ for the 2,4,6-TCP adsorption onto GO-Fe3O4@P have been estimated, suggesting the adsorption process was endothermic and entropy favored in nature.
 Please wait while we load your content...
Please wait while we load your content...

