
One-pot synthesis of Pd@PdPt core–shell nanocubes on carbon supports
Cheonghee Kimab,
Jiwhan Kimc,
Sungeun Yanga and
Hyunjoo Lee*c
aDepartment of Chemical and Biomolecular Engineering, Yonsei University, Seoul 120-749, South Korea
bClean Energy Research Center, Korea Institute of Science and Technology, Seoul 136-791, South Korea
cDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology, Daejeon 305-701, South Korea. E-mail: azhyun@kaist.ac.kr
First published on 18th November 2014
Abstract
Cubic Pd@PdPt core–shell nanoparticles were synthesized on carbon supports using a one-pot process. The as-prepared cubic Pd@PdPt/C exhibited high electrocatalytic activity for oxygen reduction reactions. The enhancement in the mass activity increased after accelerated durability tests due to structural rearrangement.
Pt-based bimetallic nanoparticles have been reported to have improved catalytic activity and contribute toward reducing the Pt cost for various fuel cell reactions, such as oxygen reduction reactions (ORRs).1–4 Among the various metals, Pd has been considered a promising co-catalyst due to the same face-centered cubic structure and similar lattice constants as Pt.5 Shape effects of metallic nanoparticles have been reported for Pt and Pd. Pt cubes with a Pt(100) surface showed higher ORR activity than did spherical Pt nanoparticles.6 Similarly, Pd cubes with a Pd(100) surface also presented higher ORR activity than did spherical Pd nanoparticles.7
Shaped metallic nanoparticles have different surface crystalline structures with various atomic arrangements. The catalytic activity, selectivity, and durability can be tuned by modulating the morphology of the metallic nanoparticles.8–13 The shape-control of nanoparticles is typically conducted by using surface-capping agents, such as polyvinylpyrrolidone or oleylamine in wet chemistry.14–16 The surface-capping agents ensure the stabilization of the colloidal nanoparticles in solution and often control the direction of overgrowth on the nanocrystal nuclei inducing the particular final shape. However, the surface-capping agents are often too strongly bound to the metal surface and block the catalytically active sites. Their removal prior to catalytic reactions is very important; however, the particular shape is often destroyed during the removal process.17–19 Previously, we showed that Pt cubes could be synthesized directly on the carbon support without using surface-capping agents.20
Many types of Pt–Pd bimetallic nanoparticles have been prepared using surface-capping agents.21–25 Here, we designed a one-pot synthesis of cubic shaped Pt–Pd bimetallic nanoparticles with Pd cores and Pt–Pd shells on carbon supports (denoted as Pd@PtPd/C) without surface-capping agents; the synthesized nanoparticles were tested as electrocatalysts for ORR in an alkaline solution.
The cubic Pd@PdPt/C core–shell catalysts were synthesized in one pot using Pd and Pt precursors, carbon black, and cysteamine in octadecene. The TEM image in Fig. 1(a) clearly shows that cubic nanoparticles were obtained after the synthesis. The average particle size was 7.1 ± 1.2 nm. A high-resolution TEM image (Fig. 1(b)) and its fast Fourier-transform image confirmed the cubic shape. The core–shell structure of the Pd@PdPt nanoparticle was confirmed by a cross-sectional compositional line scan profile measured using HAADF-STEM; Fig. 1(c) clearly shows that the core consists of Pd mainly and that the shell is an alloy of Pd and Pt with more Pt. The core–shell structure was further confirmed by separately measuring the bulk and surface compositions using ICP and XPS, respectively. The atomic ratio of Pt/(Pd + Pt) was 0.41 and 0.48 for ICP and XPS measurements, respectively. This finding indicates that Pt was located on the surface more. XRD data in Fig. 1(d) presents that these nanoparticles are truly alloys, not a binary mixture of separate Pd and Pt nanoparticles.
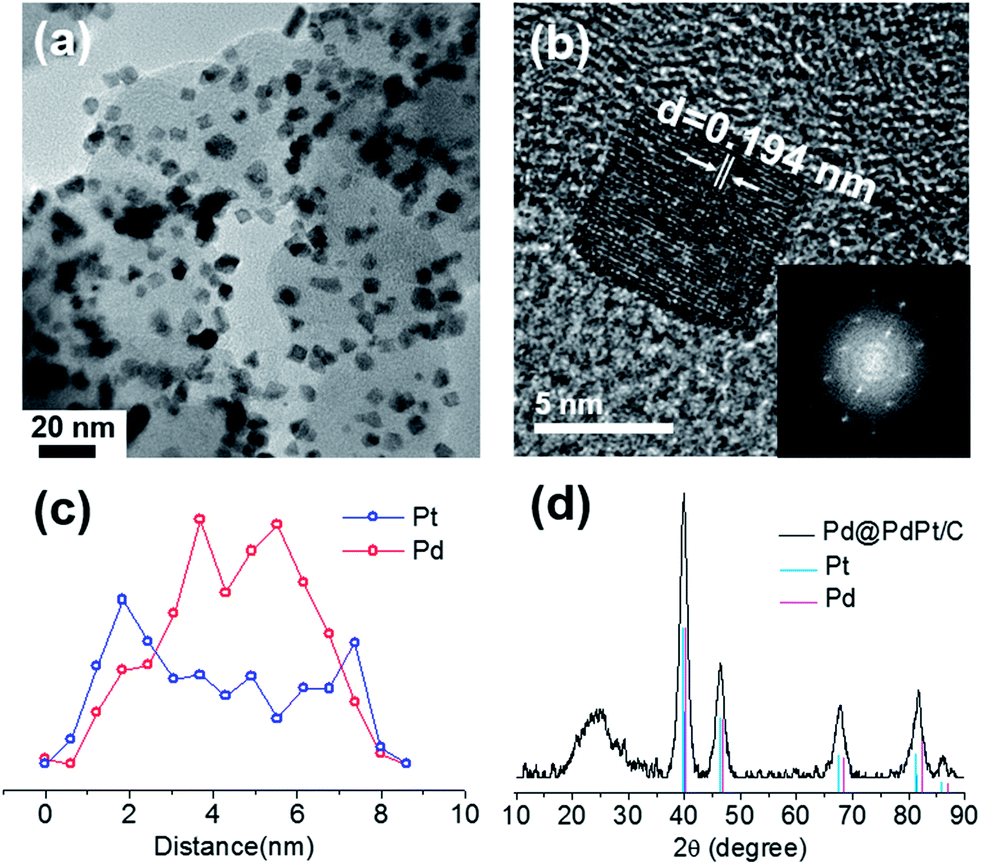 | ||
| Fig. 1 (a) TEM image of the cubic Pd@PdPt/C. (b) A high resolution TEM image presenting lattices (an inset shows its fast Fourier-transformed image showing the cubic shape clearly). (c) A cross-sectional compositional line profile of a cubic Pd@PdPt nanoparticle measured by HAADF-STEM. (d) XRD data of the cubic Pt@PdPt nanoparticles with crystalline peaks for Pd and Pt only. | ||
When TEM images were taken for the nanoparticles obtained without washing, isolated unsupported nanoparticles were not observed. Cysteamine (SHCH2CH2NH2) molecules with thiol and amine end-groups acted as anchoring agents to initiate direct nucleation on the carbon support. When cysteamine was mixed with carbon black by ultrasonication, it was immobilized on the carbon support.20 The thiol group was detected by XPS after thorough washing. Then, the metal precursors were reduced in the 1-octadecene solution by increasing the temperature. When Pd precursor was reduced in 1-octadecene solution without Pt precursor, it started to be reduced at 160 °C. On the other hand, Pt precursor alone started to be reduced at 200 °C. When the mixture of Pd and Pt precursors were reduced by increasing the reaction temperature, Pd first formed nuclei on the carbon support, and Pt later formed the shell at a higher temperature. Fig. 2(a) shows the TEM image of a sample taken from the flask at 180 °C. Pd nuclei with 2–3 nm in size were observed. No floating nanoparticles were observed, indicating that the nucleation occurred at the carbon surface. When the temperature approached 230 °C, the particle size increased, and a cubic structure appeared. After 60 min at 230 °C, the nanoparticles were fully developed. The thiol group in cysteamine appeared to cause the cubic shape. H2S and NaSH have been shown to make Pt and Ag cubes, respectively.26,27
 | ||
| Fig. 2 TEM images of the as-made cubic Pd@PdPt/C measured without a washing process when the reaction temperature approached (a) 180 °C and (b) 230 °C. | ||
The prepared cubic Pd@PdPt/C were used as catalysts for an electrochemical reaction. The ORR was performed in alkaline solution, and the catalytic activity and durability were compared with a commercial Johnson-Matthey Pt/C. The Pd@PdPt/C presented electronic signals without any particular post-treatment. Typically, shaped metallic nanoparticles synthesized with surface-capping agents require harsh post-treatments such as imposing high voltages to remove the organic layer from the metal surface. However, the shaped alloy nanoparticles we prepared here were clean enough to present electronic signals after thorough washing, thus they need not be cleaned further. The durability for ORR was tested by cycling the potential in the range of −0.3–0.1 V (vs. Ag/AgCl). Fig. 3(a) shows the activity at 0.9 V (vs. RHE) of commercial Pt/C and Pd@PdPt/C catalysts at the initial stage and after running cyclic voltammograms (CV) for 5000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. The initial mass activities at 0.9 V (vs. RHE) were similar for the two catalysts: 0.106 mA μgmetal−1 for Pd@PdPt/C and 0.107 mA μgPt−1 for commercial Pt/C. Whereas the mass activity decreased to 0.076 and 0.048 mA μgPt−1 for commercial Pt/C, however, the mass activity of Pd@PdPt/C increased to 0.138 and 0.182 mA μgmetal−1 after 5000 and 10000 cycles of CV, respectively. The mass activity of Pd@PdPt/C was 3.8 times higher than that of commercial Pt/C after 10000 cycles. The specific activity presented similar trends. Though the specific activity of commercial Pt/C decreased from 0.27 mA cmPt−2 at the initial stage to 0.17 mA cmPt−2 after 10000 cycles of ORR, the specific activity increased for Pd@PdPt/C from 0.55 mA cm−2 to 0.68 mA cm−2. Fig. 3(b) shows ORR polarization curves for both catalysts after 10000 cycles. The x-axis represents the potentials of the reversible hydrogen electrode. The ORR curve for Pd@PtPd/C is shifted more to the right than the curve of the commercial Pt/C.
000 cycles. The initial mass activities at 0.9 V (vs. RHE) were similar for the two catalysts: 0.106 mA μgmetal−1 for Pd@PdPt/C and 0.107 mA μgPt−1 for commercial Pt/C. Whereas the mass activity decreased to 0.076 and 0.048 mA μgPt−1 for commercial Pt/C, however, the mass activity of Pd@PdPt/C increased to 0.138 and 0.182 mA μgmetal−1 after 5000 and 10000 cycles of CV, respectively. The mass activity of Pd@PdPt/C was 3.8 times higher than that of commercial Pt/C after 10000 cycles. The specific activity presented similar trends. Though the specific activity of commercial Pt/C decreased from 0.27 mA cmPt−2 at the initial stage to 0.17 mA cmPt−2 after 10000 cycles of ORR, the specific activity increased for Pd@PdPt/C from 0.55 mA cm−2 to 0.68 mA cm−2. Fig. 3(b) shows ORR polarization curves for both catalysts after 10000 cycles. The x-axis represents the potentials of the reversible hydrogen electrode. The ORR curve for Pd@PtPd/C is shifted more to the right than the curve of the commercial Pt/C.
 | ||
| Fig. 3 (a) The mass activity and specific activity of commercial Pt/C and cubic Pd@PdPt/C at a fixed potential of 0.9 V (vs. RHE) before and after accelerated ORR durability tests. (b) ORR polarization curves for commercial Pt/C and cubic Pd@PdPt/C after 10000 cycles of accelerated ORR durability tests. Cyclic voltammograms measured in 0.1 M KOH solution after accelerated ORR durability tests for (c) commercial Pt/C and (d) cubic Pd@PdPt/C. | ||
The CVs in 0.1 M KOH solution before and after the accelerated ORR durability tests are shown in Fig. 3(c) and (d). The differences indicate that cubic Pd@PdPt/C has a superior long-term stability relative to commercial Pt/C. Whereas the ECSA for commercial Pt/C declined 17% and 30% after 5000 and 10000 cycles, respectively, the ECSA of cubic Pd@PdPt/C increased 30% and 38% after 5000 and 10000 cycles. Fig. 4 shows TEM images of the catalysts after the durability test. As observed, the cubic Pd@PdPt/C has minor shape deformations, whereas the commercial Pt/C shows severe aggregation. Anchoring to the carbon support minimized aggregation for the cubic Pd@PdPt/C. Furthermore, the H adsorption/desorption peaks and shoulders were shifted to the right for the cubic Pd@PdPt/C after the accelerated durability test. The H desorption peak was initially at 0.27 V but shifted to 0.30 V after 10000 cycles. This shift indicates that the composition of the active surface might have changed.22,28 After 10000 cycles, when the cubic Pd@PdPt/C was observed by cross-sectional compositional line scans using HAADF-STEM, as shown in Fig. 4(c), the overall distribution was different from the profile shown in Fig. 1(c). Fewer Pd was observed on the shell, and more Pd was found concentrated in the core. The atomic ratio of Pt/(Pd + Pt), as determined by XPS, also increased from 0.48 to 0.54. After the durability test, the shell seemed to have an increased amount of Pt due to structural rearrangement. The mesoporous carbon support with higher surface area can be used for the heavier deposition of shaped metal nanoparticles.29,30
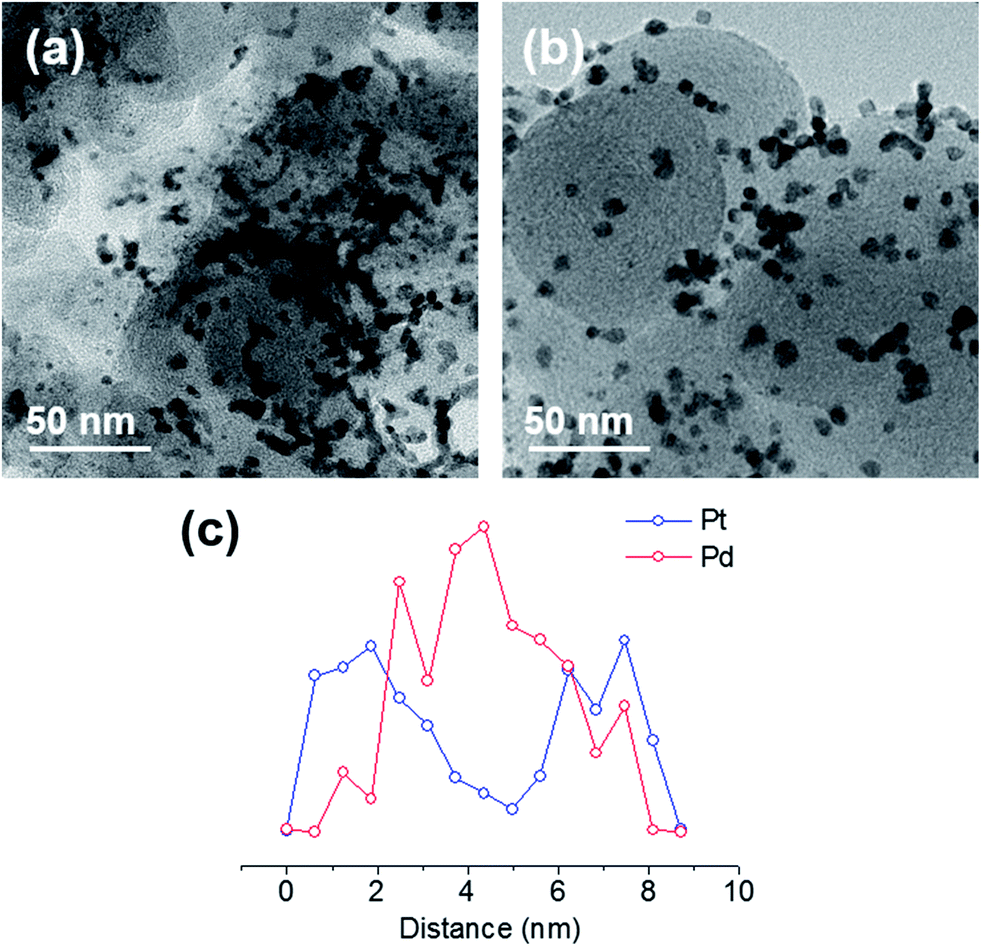 | ||
| Fig. 4 TEM images of (a) commercial Pt/C and (b) cubic Pd@PdPt/C, and (c) a cross-sectional compositional line profile of the cubic Pd@PdPt/C measured by HAADF-STEM after 10000 cycles of accelerated ORR durability tests. | ||
Experimental section
Synthesis of Pd@PdPt nanocubes on carbon supports
Ten milligrams of palladium(II) acetylacetonate (Pd(acac)2; Aldrich, 99%) and 10 mg of platinum(II) acetylacetonate (Pt(acac)2; Aldrich, 97%) were mixed with 10 mL of 1-octadecene (ODE, Aldrich) under a nitrogen flow with vigorous stirring. This solution was slowly heated to 100 °C over 25 min. Twenty milligrams of carbon black (Vulcan® XC72R) was dispersed in a mixture of 9 mL of 1-octadecene and 1 mL of cysteamine (Aldrich, 95%) by ultrasonication for 30 min. The prepared carbon solution was added to the metal precursor solution at 100 °C. The mixed solution was kept at 100 °C for 10 min. Then, the temperature was increased to 230 °C at a rate of 3–4 °C min−1 and kept at 230 °C for 60 min. The product solution was cooled down to room temperature, washed with isopropanol several times, filtered, and dried. The prepared catalysts were analyzed by X-ray photoelectron spectroscopy (XPS; Thermo Scientific K-Alpha), X-ray diffractometer (XRD; Shimadzu XRD-6000), transmission electron microscopy (TEM; JEM-2100 at 200 kV), high-angle annular dark field scanning TEM (HAADF-STEM; TECNAI F20 G2 and JEM-ARM200F at 200 kV), and energy dispersive X-ray spectroscopy (EDS; JEM-ARM200F at 200 kV).Electrochemical measurements
The catalysts were dispersed into a mixture of deionized water (0.5 mL) and isopropanol (1.5 mL) by sonication for 20 min. The actual metal content in the solution was measured by inductively coupled plasma (ICP) elemental analysis. An ink solution was dropped onto a glassy carbon rotating ring disk electrode (RRDE; Pine, 0.247 cm2). The mass of loaded metal was 3 μg for all cases. After drying, the electrode was coated with 10 μL of Nafion solution (Nafion (5 wt% solution, Aldrich):isopropanol = 1:99). The catalytic properties of the prepared cubic Pd@PdPt/C were compared with commercial Pt/C (Johnson Matthey; 20 wt% Pt/C). A platinum wire and Ag/AgCl (saturated with KCl) were used as a counter electrode and a reference electrode, respectively. Electrochemical measurements were performed using a CHI 760 potentiostat. The electrolyte solutions (0.1 M KOH) were purged with high purity N2 gas for 30 min. To obtain stable electrochemical signals, cyclic voltammetry was performed from −0.8 V to 0.2 V (vs. Ag/AgCl) for dozens of cycles prior to the measurements. The hydrogen adsorption/desorption voltammograms were acquired by cycling the potential at a scan rate of 50 mV s−1 from −0.9 V to 0.15 V (vs. Ag/AgCl). The ORR measurements were performed in an oxygen-saturated 0.1 M KOH solution at a rotating rate of 1600 rpm and a sweep rate of 10 mV s−1. To evaluate the mass and specific activities, the kinetic currents (ik) were calculated using the Koutecky–Levich equation, where i is the measured current density and id is the diffusion-limited current density. The kinetic current (ik = (i × id)/(id − i)) is independent of diffusion and is used to evaluate the intrinsic activity of the catalysts. A durability test was performed in an oxygen-saturated 0.1 M KOH solution by repeating cyclic voltammetry from −0.3 V to 0.1 V (vs. Ag/AgCl) at a scan rate of 100 mV s−1 for 5000 or 10000 cycles. The potentials were reported versus a reversible hydrogen electrode (RHE).| E(vs. RHE) = E(vs. Ag/AgCl) + 0.196 V + 0.0591 V × pH |
Conclusions
Cubic-shaped Pd@PdPt core–shell nanoparticles were directly synthesized on a carbon support using a facile one-pot method. Cysteamine added during the synthesis acted as an anchoring agent, which initiated the nucleation on the carbon support, and also acted as a shaping agent inducing the cubic shape. When Pd and Pt precursors were present together in the reacting solution, Pd was first reduced to form the nuclei followed by Pt reduction to form the shells, resulting in Pd@PdPt alloy nanoparticles. The prepared Pd@PdPt/C was applied as electrocatalysts for ORRs in alkaline media. The mass activity of Pd@PdPt/C was 3.8 times higher than that of commercial Pt/C after 10000 cycles of accelerated durability tests. Whereas both the mass and the specific activity decreased over the repeated ORRs for commercial Pt/C, the mass and the specific activity increased for Pd@PdPt/C over the reactions. The shell composition changed having more Pt after the durability test, while preserving the cubic shape.
Acknowledgements
This work was supported by the Global Frontier R&D Program of the Center for Multiscale Energy System (2011-0031575) through the National Research Foundation of Korea, and the Yonsei University Research Fund of 2013.Notes and references
- M. Shao, K. Shoemaker, A. Peles, K. Kaneko and L. Protsailo, J. Am. Chem. Soc., 2010, 132, 9253–9255 CrossRef CAS PubMed.
- J. Wu, J. Zhang, Z. Peng, S. Yang, F. T. Wagner and H. Yang, J. Am. Chem. Soc., 2010, 132, 4984–4985 CrossRef CAS PubMed.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 CAS.
- J. W. Hong, S. W. Kang, B.-S. Choi, D. Kim, S. B. Lee and S. W. Han, ACS Nano, 2012, 6, 2410–2419 CrossRef CAS PubMed.
- E. Antolini, Energy Environ. Sci., 2009, 2, 915–931 CAS.
- C. Wang, H. Daimon, T. Onodera, T. Koda and S. H. Sun, Angew. Chem., Int. Ed., 2008, 47, 3644–3647 CrossRef.
- H. Erikson, A. Sarapuu, N. Alexeyeva, K. Tammeveski, J. Solla-Gullón and J. M. Feliu, Electrochim. Acta, 2012, 59, 329–335 CrossRef CAS PubMed.
- G. A. Somorjai and D. W. Blakely, Nature, 1975, 258, 580–583 CrossRef CAS.
- F. Zaera and G. A. Somorjai, J. Am. Chem. Soc., 1984, 106, 2288–2293 CrossRef CAS.
- C. Kim and H. Lee, Catal. Commun., 2009, 11, 7–10 CrossRef CAS PubMed.
- M. Min, C. Kim, Y. I. Yang, J. Yi and H. Lee, Phys. Chem. Chem. Phys., 2009, 11, 9759–9765 RSC.
- H. Lee, S. E. Habas, S. Kweskin, D. Butcher, G. A. Somorjai and P. Yang, Angew. Chem., Int. Ed., 2006, 45, 7824–7828 CrossRef CAS PubMed.
- H. Lee, RSC Adv., 2014, 4, 41017–41027 RSC.
- H. Song, F. Kim, S. Connor, G. A. Somorjai and P. Yang, J. Phys. Chem. B, 2004, 109, 188–193 CrossRef PubMed.
- C. Wang, H. Daimon, Y. Lee, J. Kim and S. Sun, J. Am. Chem. Soc., 2007, 129, 6974–6975 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- C. Kim and H. Lee, Catal. Commun., 2009, 10, 1305–1309 CrossRef CAS PubMed.
- A. Quintanilla, V. C. L. Butselaar-Orthlieb, C. Kwakernaak, W. G. Sloof, M. T. Kreutzer and F. Kapteijn, J. Catal., 2010, 271, 104–114 CrossRef CAS PubMed.
- I. Lee, R. Morales, M. A. Albiter and F. Zaera, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15241–15246 CrossRef CAS PubMed.
- C. Kim, S. S. Kim, S. Yang, J. W. Han and H. Lee, Chem. Commun., 2012, 48, 6396–6398 RSC.
- Q. Yuan, Z. Zhou, J. Zhuang and X. Wang, Chem. Commun., 2010, 46, 1491–1493 RSC.
- H. Zhang, Y. Yin, Y. Hu, C. Li, P. Wu, S. Wei and C. Cai, J. Phys. Chem. C, 2010, 114, 11861–11867 CAS.
- A.-X. Yin, X.-Q. Min, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 3816–3819 CrossRef CAS PubMed.
- H. Zhang, M. Jin and Y. Xia, Chem. Soc. Rev., 2012, 41, 8035–8049 RSC.
- M. Shao, G. He, A. Peles, J. H. Odell, J. Zeng, D. Su, J. Tao, T. Yu, Y. Zhu and Y. Xia, Chem. Commun., 2013, 49, 9030–9032 RSC.
- Q. Zhang, C. Cobley, L. Au, M. McKiernan, A. Schwartz, L. P. Wen, J. Y. Chen and Y. N. Xia, ACS Appl. Mater. Interfaces, 2009, 1, 2044–2048 CAS.
- P. J. F. Harris, Nature, 1986, 323, 792–794 CrossRef CAS.
- G. Zhang, Z.-G. Shao, W. Lu, H. Xiao, F. Xie, X. Qin, J. Li, F. Liu and B. Yi, J. Phys. Chem. C, 2013, 117, 13413–13423 CAS.
- A. Prabhu, A. Al Shoaibi and C. Srinivasakannan, Mater. Lett., 2014, 136, 81–84 CrossRef CAS PubMed.
- J. Kim, Y. S. Bae and H. Lee, Nanoscale, 2014, 6, 12540–12546 RSC.
| This journal is © The Royal Society of Chemistry 2014 |