
Secondary channels in the thermal decomposition of monomethylhydrazine (CH3NHNH2)†
Abstract
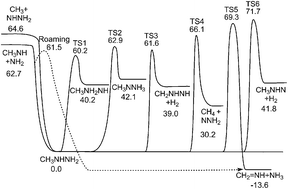
Mass spectrometric observations in a very low pressure pyrolysis study (Golden et al., Int. J. Chem. Kinet., 1972, 4, 433–448) of the decomposition of the prototypical rocket fuel monomethylhydrazine (MMH) indicated a dominant role for the molecular channels producing NH3 and H2 and their coproducts. In contrast, a recent ab initio transition state theory based master equation theoretical study (Zhang et al., Proc. Combust. Inst., 2011, 33, 425–432) indicated that simple N–N and C–N bond fissions dominate the kinetics. The possible role of molecular decomposition channels in MMH is explored further through additional investigations of the potential energy surface. These investigations consider the role of triplet channels, of roaming radical channels, and of some previously unexplored pathways for molecular decomposition. New ab initio transition state theory based master equation calculations provide revised predictions for the temperature and pressure dependence of the MMH decomposition kinetics that are in excellent agreement with recent shock tube measurements (Li et al., Comb. Flame, 2014, 161, 16–22). These calculations continue to suggest only a very limited contribution from the molecular elimination channels. A roaming pathway is suggested to provide the dominant route for direct formation of ammonia. The possible role of secondary abstraction reactions in the very-low-pressure pyrolysis experiments is briefly discussed.
 Please wait while we load your content...
Please wait while we load your content...

