
Lipid coated mesoporous silica nanoparticles as an oral delivery system for targeting and treatment of intravacuolar Salmonella infections
Abstract
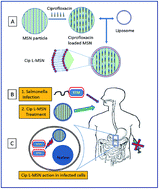
Lipid coated mesoporous silica nanoparticle (L-MSN) were synthesized for oral delivery of ciprofloxacin for intracellular elimination of Salmonella pathogen. The particle size was found to be between 50–100 nm with a lipid coat of approximately 5 nm thickness. The lipid coating was achieved by sonication of liposomes with the MSN particles and evaluated by CLSM and FTIR studies. The L-MSN particles exhibited lower cytotoxicity compared to bare MSN particles. Ciprofloxacin, a fluoroquinolone antibiotic, loaded into the L-MSN particles showed enhanced antibacterial activity against free drug in in vitro assays. The lipid coat was found to aid in intravacuolar targeting of the drug cargo as observed by confocal microscopy studies. We also observed that a lower dose of antibiotic was sufficient to clear the pathogen from mice and increase their survivability using the L-MSN oral delivery system.
 Please wait while we load your content...
Please wait while we load your content...

