
Fabrication of carbon nitride nanotubes by a simple water-induced morphological transformation process and their efficient visible-light photocatalytic activity
Abstract
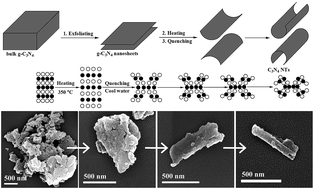
Carbon nitride nanotubes (C3N4 NTs) were synthesized based on the nanosheets roll-up mechanism by a simple water-induced morphological transformation process using graphitic carbon nitride (g-C3N4) as a precursor. Water was used as the phase-transfer reagent, making the preparation process environmentally friendly. The visible-light photocatalytic activity of the as-prepared C3N4 NTs significantly increased compared to bulk g-C3N4 and g-C3N4 nanosheets toward rhodamine B degradation and hydrogen evolution from water-splitting. This result can be attributed to the high photogenerated carrier transfer efficiency, excellent mass transfer capability, sufficient active sites, and enhanced light utilization efficiency of C3N4 NTs.
 Please wait while we load your content...
Please wait while we load your content...

