
Making a better organic–inorganic composite electrolyte to enhance the cycle life of lithium–sulfur batteries†
Abstract
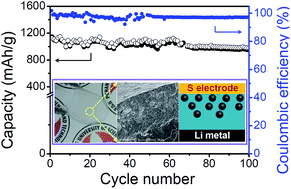
A new methodology for resolving the long-standing obstacles of Li–S batteries by the synthesis of a composite gel polymer electrolyte with a unique internal structure is disclosed in this paper. The Li–S cells prepared using this novel electrolyte system exhibit high discharge capacities (1140 mA h g−1 during the 1st cycle and reversible capacity of 970 mA h g−1 at the 100th cycle) and improved cycle life.
- This article is part of the themed collection: Polymers for Electrochemical Energy Storage
 Please wait while we load your content...
Please wait while we load your content...

