
Special Tm3+ transition and white upconversion luminescence in the Yb3+/Tm3+ co-doped KMnF3
Bing Maa,
Xiaoyang Liua,
Li Liub,
Yujia Menga,
Benxian Lia,
Xudong Zhaoa,
Peixuan Pua and
Xiaofeng Wang*a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry Jilin University, Changchun 130012, China. E-mail: wangxf103@jlu.edu.cn; Fax: +86-431-85168601; Tel: +86-431-85168601
bDepartment of Chemistry, Northeast Normal University, Changchun 130024, China
First published on 6th November 2014
Abstract
Special Tm3+ transition and a bright green light emission is first observed in the Yb3+/Tm3+ co-doped KMnF3, which are synthesized by the molten salt synthesis method. The RGB light emissions are simultaneously obtained with a single Tm3+ activator, and white upconversion luminescence is observed only by changing the reaction time.
1. Introduction
Rare-earth (RE) ion doped materials with frequency upconversion (UC) luminescence have been the focus of the related scientific research owing to their applications in solid-state blue-green lasers, three-dimensional color displays, solar cells, and fluorescent bio-labels.1–4 In particular, RE3+-doped materials capable of white UC emission have attracted enormous attention.5–10 In the field of color-tunable UC luminescence, more and more efforts are being devoted for the development of materials emitting white UC light. Generally, white light is obtained by mixing three monochromatic light sources RGB (red, green, and blue). The common method of inducing RGB light emissions is to add a variety of rare earth ions with colorful UC emissions.11–13For example, white UC luminescence is observed in the tri-doped Yb3+, Tm3+ and Er3+ system upon 980 nm diode laser excitation, which consisted of blue (1D2 → 3F4, 1G4 → 3H6 of Tm3+), green (2H11/2/4S3/2 → 4I15/2 of Er3+), and red (4F9/2 → 4I15/2 of Er3+) UC emissions.14 Even though green (1D2 → 3H5, 550 nm) and red (1G4 → 3F4, 650 nm) emissions can also be produced by the energy level transition of Tm3+, they are generally weak or even cannot be detected due to their low transition probabilities.15 Consequently, if the transition probabilities of the 1D2 and 1G4 levels of Tm3+ could be adequately altered, the RGB light emissions output from a single activator (Tm3+) would be achievable. Furthermore, white UC light can also be obtained by controlling the intensities and ratios of the RGB light emissions in the Yb3+/Tm3+ co-doped systems.
The RGB light emissions of Tm3+ are usually originated from the excitation of high energy levels (1D2 and 1G4). However, the sensitization for high energy level transition is seldom reported in the literature.16–19 Notably, the influence of the sensitizer (Mn2+) on the UC emission behaviors has been discussed in detail by Tian's group and Wang's group.16,17 The results reveal that there is an efficient energy transfer between the RE3+ (RE = Er, Tm, Ho) and Mn2+. Therefore, the increase in NIR emission transition of Tm3+ can be attributed to the non-radiative energy transfer from the 1D2 and 1G4 levels of Tm3+ to the 4T1 level of Mn2+, followed by a back-energy transfer to the 3F2,3 level of Tm3+. Because the ratios of light emissions show a linear relation with the sensitized abilities,11 it is highly possible that the intensities and ratios of RGB light emissions could be modulated by introducing the sensitizer (Mn2+) into the Yb3+/Tm3+ co-doped KMnF3 system.
To that end, we have synthesized a series of Yb3+/Tm3+ co-doped KMnF3 (KMnF3:Yb, Tm) crystals utilizing the molten salt synthesis (MSS) method, followed by a detailed study on UC luminescence properties. The research results show that the reaction time is a crucial factor to affect the UC luminescence performance of KMnF3:Yb, Tm system. It is noteworthy that a unique change has taken place in the 1D2 level transition of Tm3+ as the reaction time was extended. First, a strong green emission is observed in such materials, which is originated from the excitation of the activator (Tm3+). In addition, the novel transition process of Tm3+ has been found, which increases the low transition probability and makes it possible for obtaining all the RGB light emissions solely from the activator (Tm3+). Therefore, it appears that white UC light can be rationally designed by controlling the relative intensities of the RGB light emissions in the Yb3+/Tm3+ co-doped KMnF3 material, which could be readily achieved by varying the reaction time used. To our knowledge, it is the first example of obtaining white UC light via employing a single Tm3+ activator in the RE3+-doped luminescence materials. The change in the transition probabilities provided a new route for the color-tunable UC fluorescence. White UC light was just one aspect of the application, more potential applications will be found in the future.
2. Experimental section
All the reagents were obtained from commercial sources and directly used without purification. For a typical synthesis, KCl, MnCl2·4H2O, NH4HF2, Yb(NO3)3·5H2O, and Tm(NO3)2·6H2O were added to an autoclave in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:10:5%:0.5%. The autoclave was sealed and maintained at 180 °C for different reaction time. After naturally cooling down to room temperature, the resulting products were washed with deionized water and dried overnight at 60 °C.
:1:10:5%:0.5%. The autoclave was sealed and maintained at 180 °C for different reaction time. After naturally cooling down to room temperature, the resulting products were washed with deionized water and dried overnight at 60 °C.
All the samples were characterized by powder X-ray diffraction (XRD, Cu Kα radiation, Rigaku D/max2550VB, Japan) at a scanning rate of 8° min−1 within the 2θ range of 10° to 80°. The morphology and particle size of the samples were determined by scanning electron microscopy (SEM, JSM-6700F, JEOL, Japan). The UC emission spectra were measured on an Edinburgh Instruments FLS920 spectrophotometer with an external 0–600 mW adjustable continuous-wave 980 nm laser as the excitation source. The particle surface was characterized by X-ray photoelectron spectroscopy (XPS, Al Kα radiation, America, ESCALAB 250). All the optical measurements were performed under ambient conditions.
3. Results and discussion
The crystalline structures of the as-synthesized products are identified by powder X-ray diffraction (XRD). Fig. 1 shows the XRD patterns of KMnF3 prepared at 180 °C with different reaction times (30 min, 6 h, 12 h, and 24 h), as well as the standard XRD data of pure cubic KMnF3 phase as a reference. The XRD results show that all the samples are well crystallized. All the diffraction peaks can be indexed to the cubic phase of KMnF3 (JCPDS: 17-0116), indicating the high purity of the as-synthesized products. | ||
| Fig. 1 XRD patterns of the KMnF3 samples prepared at 180 °C with different reaction times (A) 30 min, (B) 6 h, (C) 12 h, and (D) 24 h, and the standard data of cubic KMnF3 (JCPDS: 17-0116). | ||
Fig. 2 shows the scanning electron microscopy (SEM) images of the KMnF3:Yb, Tm samples prepared at different reaction times (30 min, 6 h, 12 h, and 24 h), in which the resulting crystals are clearly cube-shaped. It is found that the size of the crystals significantly increases with the increase in reaction time. When the reaction is terminated after 30 min, the size of crystals is less than 1 μm, whereas if the reaction is allowed to proceed for 24 h, the crystal size turns out to be 5 μm.
 | ||
| Fig. 2 SEM micrographs of the Yb/Tm co-doped KMnF3 samples prepared with different reaction times (A) 30 min, (B) 6 h, (C) 12 h, (D) 24 h. | ||
The UC emission spectra of the KMnF3:Yb, Tm crystals prepared with different reaction times (30 min, 6 h, 12 h, and 24 h) are exhibited in Fig. 3. The NIR UC emission at 800 nm corresponds to the 3H4 → 3H6 transition (Fig. 3A), which is due to the excitation of the 3F2,3 level, followed by the non-radiative relaxation from the 3F2,3 level to the 3H4 level. Notably, two weak bands at 475 nm and 650 nm are observed corresponding to the 1G4 → 3H6 and 1G4 → 3F4 transition of Tm3+, respectively. In addition, no 1D2 level transition is observed, and two 1G4 level transitions (1G4 → 3H6 and 1G4 → 3F4) seem to be weak in Fig. 3A. These results could be attributed to the non-radiative energy transfer from the 1D2 and 1G4 levels of Tm3+ to the 4T1 level of Mn2+, followed by a back-energy transfer to the 3F2,3 level of Tm3+. When the reaction time is increased to 6 h, the band at 650 nm seems to be substantially enhanced, as illustrated in Fig. 3B. Further extending the reaction time to 12 h, two new bands at 450 nm and 550 nm clearly appeared, corresponding to the 1D2 → 3F4 and 1D2 → 3H5 transition of Tm3+, respectively (Fig. 3C). Therefore, we conclude that the 1D2 and 1G4 level transitions can be simply enhanced by extending the reaction time, as illustrated in Fig. 3A to C. In other words, the sensitization of Mn2+ can be substantially decreased during this process. As shown in Fig. 4, the exchange-energy transfer process from the 1D2 level of Tm3+ to the 4T1 level of Mn2+ is more efficient than the one from the 1G4 level to the 4T1 level. When the reaction time is increased to 24 h, the 1D2 level emission of Tm3+ at 550 nm is found to be stronger, whereas the band at 450 nm seems to be disappeared, as illustrated in Fig. 3D. Generally, when the 1D2 level of Tm3+ is excited, the most likely transition process should be the 1D2 → 3F4 transition at 450 nm; other probable transitions, however, would be fairly weak or even negligible due to their low transition probabilities. Surprisingly, we observe an obvious band at 550 nm (1D2 → 3H5) in the emission spectra of KMnF3:Yb, Tm samples prepared at 24 h (Fig. 3D), which has never been reported in the literature and no reliable mechanism with it. The result suggests that an unprecedented alteration has occurred in the 1D2 level transition of Tm3+. In general, 1D2 → 3F4 (450 nm, blue) transition is probability almost 220 times that of 1D2 → 3H5 (550 nm, green),15 the strong green light emission from Tm3+ was due to the change of the 1D2 level transition probabilities ratio between them. The synthesis conditions of the KMnF3 : Yb, Tm crystals in our work are try to keep the same with the traditional solid-phase synthesis except the NH4HF2 flux, the excess F− are the most probable reason, but still need more evidence to prove. Similar results can be observed in the Yb3+/Tm3+ co-doped β-NaYF4 system (Fig. 5), which has been prepared in the absence of Mn2+ by the MSS method in our research group. On the basis of the data collected from these two examples, we conclude that the abnormal change of transition probabilities could originate from the influence of the crystal structure and micrometer size, rather than the introduction of Mn2+. In addition, it is proved that the RGB light emissions can be simultaneously obtained by a single Tm3+ activator in the Yb3+/Tm3+ co-doped system, which are red (1G4 → 3F4, at 650 nm), green (1D2 → 3H5, at 550 nm), and blue (1D2 → 3F4, at 450 nm and 1G4 → 3H6, at 475 nm) UC emissions. As a matter of fact, the intensities and ratios of the RGB light emissions can be solely modulated by controlling the reaction time, affording white light emission.
 | ||
| Fig. 3 Room temperature UC emission spectra of the KMnF3:Yb, Tm samples prepared with different reaction times (A) 30 min, (B) 6 h, (C) 12 h, (D) 24 h. The samples were excited with a 980 nm laser operating at 600 mW (The insets are the magnified image for the visible light area). | ||
 | ||
| Fig. 4 Energy level diagrams of the Yb3+, Tm3+, and Mn2+, and the efficient transfer process from the 1D2 level of Tm3+. | ||
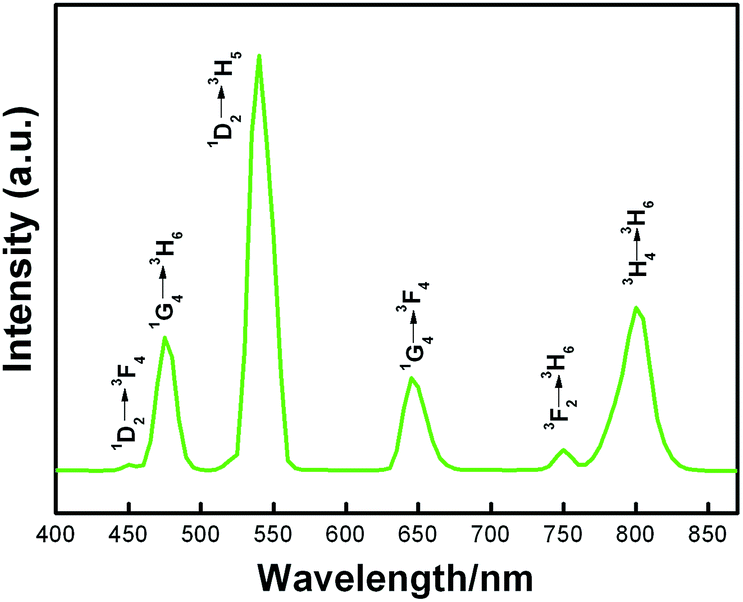 | ||
| Fig. 5 Room temperature UC emission spectra of the Yb3+/Tm3+ co-doped β-NaYF4 sample prepared at 180 °C for 24 h. The samples were excited with a 980 nm laser operating at 600 mW. | ||
All the UC emission spectra of the KMnF3:Yb, Tm crystals can be readily converted to the CIE-1931 chromaticity diagrams, as plotted in Fig. 6. The corresponding chromaticity coordinates of crystals have been calculated based on the emission spectra, which are (0.4435, 0.2428), (0.4081, 0.3216), (0.3532, 0.3499) and (0.3151, 0.6018), respectively. As shown in the chromaticity diagrams, the color tone of the crystals gradually shifts from red to white, and ultimately to green with the prolongation of the reaction time. Therefore, it appears that the white light emission can be obtained only by varying the reaction time utilized in the synthetic process. Indeed, when the reaction time is prolonged from 6 h to 12 h, the chromaticity coordinates could locate in the white light range as expected. By further extending the reaction time to 24 h, the chromaticity coordinate could move into the green light area. Differing from the previously reported color-tunable methods,20–22 the approach reported in this work can produce red, green and white fluorescent materials based on the KMnF3:Yb, Tm system by varying the reaction time, and no changes to any other reaction conditions are needed.
 | ||
| Fig. 6 CIE chromaticity diagram of the KMnF3:Yb, Tm samples prepared with different reaction times (A) 30 min, (B) 6 h, (C) 12 h, (D) 24 h. | ||
The X-ray photoelectron spectroscopy (XPS) spectrum (Fig. 7) shows that the concentration of Mn2+ on the surface of the crystal is decreased with the extension of reaction time, whereas the concentration of Yb3+ is increased. It is noted that the energy exchange interaction between Mn2+ and Tm3+ is weakened and between Yb3+ and Tm3+ is strengthened, resulting in a gradual increase in the 1D2 level transitions in Tm3+. In addition, the Yb3+/Er3+ co-doped KMnF3 crystals have also been synthesized at the same reaction condition by our research group, similar results are also obtained from the UC emission spectra, as shown in Fig. 8.
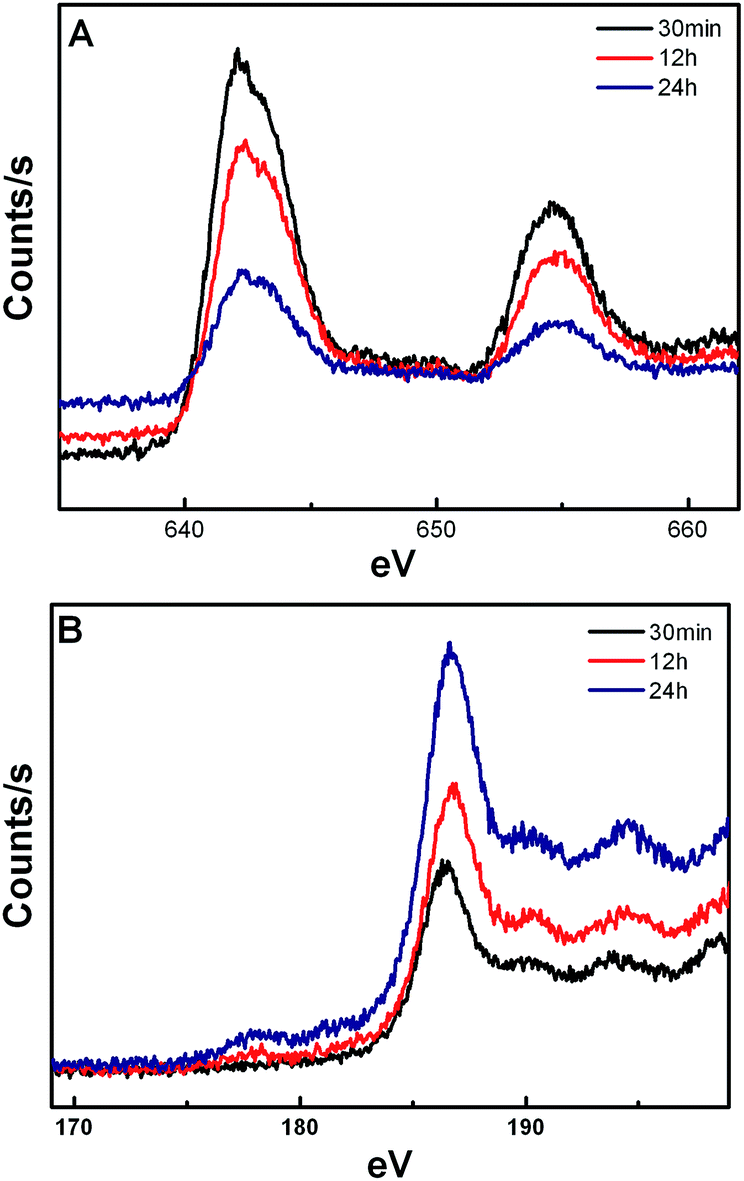 | ||
| Fig. 7 XPS spectra for the KMnF3:Yb, Tm samples (A) Mn2+ 2p; (B) Yb3+ 4d. | ||
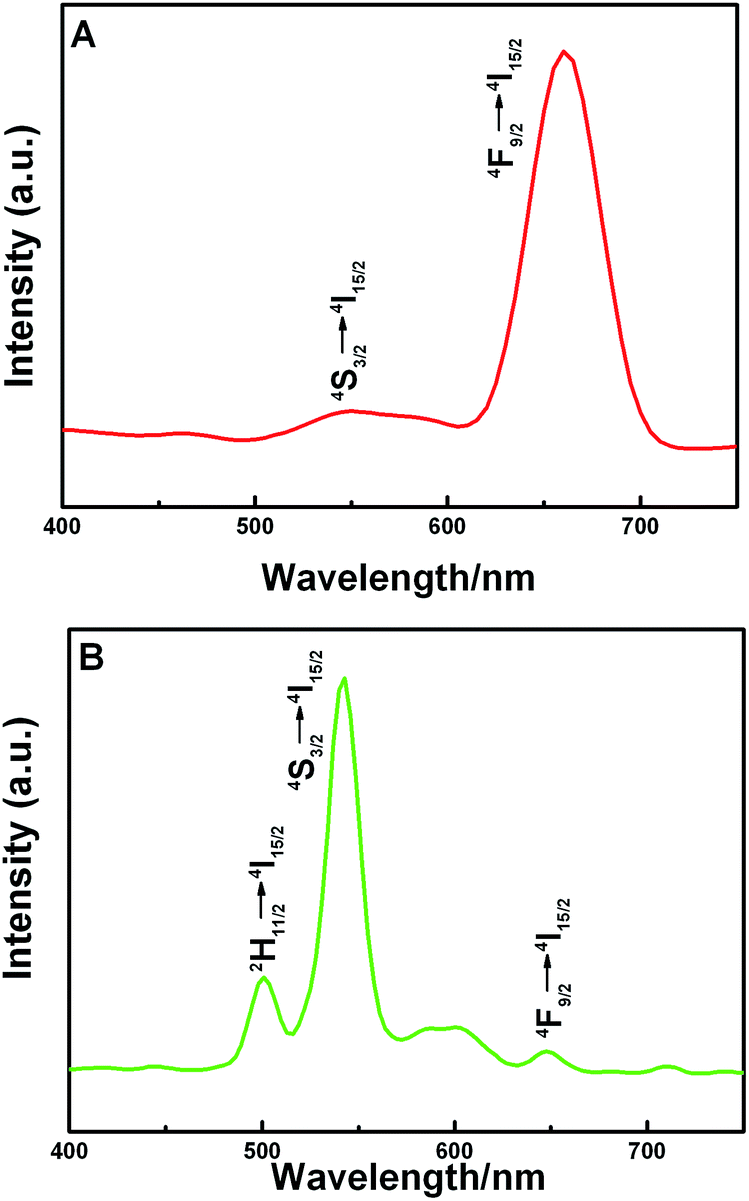 | ||
| Fig. 8 Room temperature UC emission spectra of the Yb3+/Er3+ co-doped KMnF3 samples prepared at 180 °C with different reaction times (A) 6 h, (B) 24 h. The samples were excited with a 980 nm laser operating at 600 mW. | ||
4. Conclusions
We have successfully synthesized the Yb3+/Tm3+ co-doped KMnF3 crystals by a MSS method. Subsequent studies indicate that the UC luminescence properties of such materials can be effectively modulated by varying the reaction time. Specifically, it appears that the 1D2 level transition of Tm3+ undergoes unprecedented changes while the reaction time is prolonged, which results in a bright green light emission from Tm3+ is. The RGB light emissions have been simultaneously obtained in the presence of a single activator (Tm3+). Moreover, it is found that the intensities and ratios of RGB light emissions can be controlled by varying the reaction time, producing white light emission in the KMnF3:Yb, Tm system. To our knowledge, white UC light is successfully obtained for the first time in the Yb3+/Tm3+ co-doped system. Overall, this work not only provides a new route for the development of white UC light emitting materials, but also demonstrates that the colorful emissions can be readily modulated in a broad range by varying the reaction conditions employed in the synthesis. Currently, our efforts are primarily focused on the preparation of more diversified RE3+-doped luminescence materials, and the studies to understand the intrinsic mechanism involved in the change of transition probability are still ongoing.Acknowledgements
This work was supported by the National Sciences Foundation of China (no. 21271082 and 21301066).References
- F. Auzel, Chem. Rev., 2004, 104, 139 CrossRef CAS PubMed.
- E. Downing, L. Hesselink, J. Ralston and R. Macfarlance, Science, 1996, 273, 1185 CAS.
- E. D. Rosa, P. Salas, H. Desirenca, C. Angeles and R. A. Rodríguez, Appl. Phys. Lett., 2005, 87, 241912 CrossRef PubMed.
- F. Van de Rijke, H. Zijlmans, S. Li, T. Vail, A. K. Raap, R. S. Niedbala and H. J. Tanke, Nat. Biotechnol., 2001, 19, 273 CrossRef CAS PubMed.
- M. S. Wang, S. P. Guo, Y. Li, L. Z. Cai, J. P. Zou, G. Xu, W. W. Zhou, F. K. Zheng and G. C. Guo, J. Am. Chem. Soc., 2009, 131, 13572 CrossRef CAS PubMed.
- N. Guo, H. P. You, Y. H. Song, M. Yang, K. Liu, Y. J. Huang and H. J. Zhang, J. Mater. Chem., 2010, 20, 9061 RSC.
- K. Z. Zheng, D. S. Zhang, D. Zhao, N. Liu, F. Shi and W. P. Qin, Phys. Chem. Chem. Phys., 2010, 12, 7620 RSC.
- T. Passuello, F. Piccinelli, M. Pedroni, M. Brttinelli, F. Mangiarini, R. Naccache, F. Vetrone, J. A. Capobianco and A. Speghini, Opt. Mater., 2011, 33, 643 CrossRef CAS PubMed.
- C. M. Zhang, P. A. Ma, C. X. Li, G. G. Li, S. S. Huang, D. M. Yang, M. M. Shang, X. J. Kang and J. Lin, J. Mater. Chem., 2011, 21, 717 RSC.
- M. M. Shang, C. X. Li and J. Lin, Chem. Soc. Rev., 2014, 43, 1372 RSC.
- J. Wang, R. R. Deng, M. A. MacDonald, B. Chen, J. K. Yuan, F. Wang, D. Z. Chi, T. S. A. Hor, P. Zhang, G. K. Liu, Y. Han and X. G. Liu, Nat. Mater., 2013, 13, 157 CrossRef PubMed.
- E. M. Chen, G. Han, J. D. Goldberg, D. J. Gargas, A. D. O. P. J. Schuck, B. E. Cohen and D. J. Milliron, Nano Lett., 2012, 12, 383 CrossRef PubMed.
- F. Wang and X. G. Liu, J. Am. Chem. Soc., 2009, 130, 5642 CrossRef PubMed.
- K. Z. Zhang, D. S. Zhang, D. Zhao, N. Liu, F. Shi and W. P. Qin, Phys. Chem. Chem. Phys., 2010, 12, 7620 RSC.
- G. Poirier, V. A. Jerez, C. B. de Araújo, Y. Messaddeq, S. J. L. Ribeiro and M. Poulain, J. Appl. Phys., 2003, 93, 1493 CrossRef CAS PubMed.
- G. Tian, Z. J. Gu, L. J. Zhou, W. Y. Yin, X. X. Liu, L. Yan, S. Jin, W. L. Ren, G. M. Xing, S. J. Li and Y. L. Zhao, Adv. Mater., 2012, 24, 1226 CrossRef CAS PubMed.
- J. Wang, F. Wang, C. Wang, Z. Liu and X. G. Liu, Angew. Chem., Int. Ed., 2011, 50, 10369 CrossRef CAS PubMed.
- N. Guo, H. P. You, Y. H. Song, M. Yang, K. Liu, Y. H. Zheng, Y. J. Huang and H. J. Zhang, J. Mater. Chem., 2010, 20, 9061 RSC.
- J. Yang, C. M. Zhang, C. X. Li, Y. N. Yu and J. Lin, Inorg. Chem., 2008, 47, 7262 CrossRef CAS PubMed.
- J. Yang, C. M. Zhang, C. Peng, C. X. Li, L. L. Wang, R. T. Chai and J. Lin, Chem.–Eur. J., 2009, 15, 4649 CrossRef CAS PubMed.
- K. Z. Zheng, D. S. Zhang, D. Zhao, N. Liu, F. Shi and W. P. Qin, Phys. Chem. Chem. Phys., 2010, 12, 7620 RSC.
- J. W. Wang and P. A. Tanner, J. Am. Chem. Soc., 2010, 132, 947 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |