
Centrifugal partition chromatography: an efficient tool to access highly polar and unstable synthetic compounds on a large scale†
Abstract
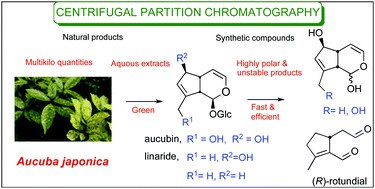
Centrifugal partition chromatography (CPC) is an efficient method adaptable for the purification of synthetic compounds with chemical value, significantly simplifying the reaction work-up. From Aucuba japonica Thunb., an abundant natural terpenoid, aucubin, was isolated. Its reduced derivatives and corresponding unstable aglucons were purified by CPC. The latter chiral synthons were obtained under protecting group free conditions using eco-friendly, non-chlorinated solvents and without silica gel.
 Please wait while we load your content...
Please wait while we load your content...

