
Fullerene modification of gold electrodes and gold nanoparticles based on application of aromatic thioacetate-functionalized C60†
Abstract
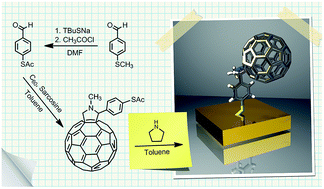
A new procedure for the synthesis of S-acetyl-derivatized fullerene is described. The deprotected aromatic S-acetyl-derivatized fullerene was employed for efficient modification of a gold electrode and gold nanoparticles. The proposed deposition procedure of fullerene derivative on Au surfaces allows the control of the thickness of the self-assembled fullerene layer. The modified surfaces were characterized by electrochemical methods and X-ray photoelectron spectroscopy (XPS). Functionalized fullerenes largely retain favorable redox electronic properties, behaving as an electron sink and revealing 4 reversible sequential 1e electrode processes at slightly more negative potentials than those observed for unsubstituted C60. For the first time, fullerene-capped gold nanoparticles were obtained by a two-step ligand exchange procedure involving substitution of alkanethiol with thiolated fullerene derivative following the synthesis of alkanethiol capped nanoparticles. The ligand exchange procedure was very efficient – the number of fullerene moieties per single gold nanoparticle was found to be as high as 30.
 Please wait while we load your content...
Please wait while we load your content...

