
A time dependent DFT study of the efficiency of polymers for organic photovoltaics at the interface with PCBM†
N. Van den Brandeab,
G. Van Liera,
F. Da Pieve‡
a,
G. Van Asscheb,
B. Van Meleb,
F. De Profta and
P. Geerlings*a
aGeneral Chemistry (ALGC), Vrije Universiteit Brussel (VUB), Pleinlaan 2, B-1050 Brussels, Belgium. E-mail: pgeerlin@vub.ac.be
bPhysical Chemistry and Polymer Science (FYSC), Vrije Universiteit Brussel (VUB), Pleinlaan 2, B-1050 Brussels, Belgium
First published on 15th October 2014
Abstract
The interface between donor and acceptor material in organic photovoltaics is of major importance for the functioning of such devices. In this work, the singlet excitation schemes of six polymers used in organic photovoltaics (P3HT, MDMO-PPV, PCDTBT, PCPDTBT, APFO3 and TBDTTPD) at the interface with a PCBM acceptor were studied using TDDFT in combination with the range-separated CAM-B3LYP exchange–correlation functional. By comparing with the excitations in the pure polymer and analyzing the excitation intensities and a measure for orbital overlap, it was possible to identify excitations as either excitation of the polymer or as a charge transfer between donor and acceptor. By combining orbital overlaps between the molecular orbitals involved in charge transfer and the intensity of the polymer excitation a broad correlation was seen with the record efficiencies found in the literature.
Introduction
Organic photovoltaics (OPVs) are considered a promising alternative for fossil fuels, due to their many advantages such as easy processability and flexibility.1,2 Their main drawback however when compared to inorganic photovoltaics is the low dielectric constant of organic materials. Because of this the electron–hole pairs or excitons that are formed by excitation through light are strongly bound (>kT at room temperature), and can only dissociate or separate at the interface between donor and acceptor material due to discontinuities in the electron affinity and ionization potential.3,4Among the different types of OPVs, polymer based types are one of the most developed alternatives. In this type of OPVs, the bulk-heterojunction (BHJ) concept is often employed to increase efficiencies, which introduces several processing challenges.5,6 In the BHJ concept the polymer donor and the acceptor form a phase-separated co-continuous morphology, increasing the interface between the two components and thus also the charge transfer.7 Nonetheless, the more rudimentary bilayer devices, where a layer of donor is combined with a layer of acceptor to form a smaller interface than in a BHJ device, are still developed further as well.8,9 The interface region, where donor and acceptor molecules come into contact, is thus of great importance to the global functioning of any OPV device, because it is the site of the vital charge separation required for the functioning of an OPV device. It is also the site where geminate recombination can take place, the event where an already separated electron and hole recombine, which is considered as a major loss mechanism for charge generation.10,11
Many types of conjugated polymers have been used as donor materials, of which perhaps the most widespread is poly(3-hexyl thiophene) or P3HT. A major drawback of this polymer is its limited absorption, leading to less current generation. More recently the trend has been to use donor–acceptor copolymers that have been especially designed for a lower bandgap and thus a better absorption in the visible spectrum, by incorporating electron-deficient and electron-rich building blocks on the same backbone.12,13 A general feature of all these donor materials is their conjugated backbone, combined with several mostly aliphatic sidegroups in order to improve processability. The most widely used acceptor material is [6,6]-phenyl-C61-butyric acid methyl ester (PCBM),14 which has been shown to lead to the highest efficiencies in most donor–acceptor combinations. It should be noted however that for some of the more recent donor polymers better results were achieved using the C70 analogue of PCBM.15
Following previous work in this field,16 the goal here is to provide a deeper understanding of the electronic properties of several donor–PCBM combinations at the interface using ab initio TDDFT calculations of excited states, and to attempt to develop an intuitive qualitative model for the charge transfer which takes place there. This proposed model will then be compared to experimental efficiency results. It should be stressed that due to the large size of the systems involved in this study, the methodology employed is approximate and is only expected to yield relative trends.
Methodological aspects and theoretical strategy
Studies regarding charge transfer often employ Marcus theory, which requires a great deal of theoretical formalism, being less intuitive.17,18 While theoretical (TD-DFT) investigations of OPV polymers and especially small molecules are often performed in order to calculate the bandgap or absorption spectra, the presence of the acceptor is usually not taken into account due to system size.19–22 As charge transfer in OPV devices takes place at the donor–acceptor interface, an investigation of the excitations present at this interface is required. However, the structures involved in a theoretical description of polymer OPVs are prohibitively large for high level calculations. Some conventional DFT studies at the interface have been performed,23,24 indicating the importance of orbital overlap in charge transfer and thus current generation in the P3HT/C60 system. Here a large emphasis is placed on the formation of a bridge state between donor and acceptor, making charge transfer possible. In order to further the quest for a better understanding of these phenomena, a study of several donor polymer/PCBM combinations is undertaken here using density functional theory (DFT) and time dependent density functional theory (TDDFT) quantumchemical calculations.Several difficulties arise however when TDDFT is used to describe charge transfer excitations due to the small orbital overlaps involved.25 An approach that increases the accuracy of TDDFT for such transitions is the use of a greater amount of exact exchange, which can be incorporated by using specific functionals.26 This approach will also be used in this work. The excitations calculated from TDDFT are analyzed in terms of oscillator strength and orbital overlaps. For this reason the Λ parameter is calculated, which was introduced by the group of Tozer et al. as a measure of the spatial overlap between the occupied and virtual orbitals involved in an excitation.26,27 This quantity can thus be used to characterize excitations as either low-overlap/long-distance excitations (low Λ) or high-overlap/short-distance excitations (high Λ). Charge transfer excitations, as a transfer of electrons between separate molecules, fall in the former category.
For this study, a group of five donor polymers were chosen: two conventional conjugated polymers, the benchmark polymer P3HT and MDMO-PPV as well as four polymers belonging to the donor–acceptor group: the commercial PCDTBT and PCPDTBT polymers, APFO3,28 the simplest of the APFO family and PBDTTPD, a non-commercial polymer based on thieno[3,4-c]pyrrole-4,6-dione found in literature.29 P3HT/PCBM based OPVs reached efficiencies of about 5% several years ago, explaining the role of P3HT as a benchmark material.30 More recent studies have improved this efficiency to about 7% by moving from a binary system to more complex active layers, showing that this system is certainly still relevant.31 MDMO-PPV is perhaps the oldest material studied here, and included as a reference. Efficiencies of around 2.5% have been reported for the MDMO-PPV/PCBM blend.1,32 PCDTBT based OPVs have shown efficiencies up to 7.5%, although this is in combination with the C70 analogue of PCBM.33 PCPDTBT, which was developed as a lower bandgap alternative for PCDTBT, has so far exhibited efficiencies of above 5%, although this required an additive and the acceptor was also PC71BM.34,35 The APFO3/PCBM system, being based on a non-commercial polymer, has received less attention in literature, but has been known to deliver an efficiency of about 3.5%.36 This is also the case for PBDTTPD, although the highest efficiency found in literature for PBDTTPD/PCBM is a promising 6.8%.37 The main difference between PCBM and its C70 analogue is that the C70 analogue shows a greater absorption in the visible region, leading to a higher light absorption by the acceptor.38,39 As excitation of the acceptor is not considered here, both PCBM and PC71BM based results will be used for comparison.
All ground state DFT calculations were performed in the gas phase. Geometrical optimizations were performed by using the PBE0 exchange–correlation functional40 in combination with the 3-21G* basis set.41 It was found that the amount of exact exchange is of major importance for the resulting geometry.42 PBE0 contains 25% of exact exchange, which is recommended for the geometry optimization of most semiconducting polymers according to Jacquemin et al.43 The Gaussian 09 software package was used for all geometry optimizations, as well as to visualize the molecular orbitals of the systems in their ground state.44 TDDFT calculations to elucidate the nature of the electronic excitations of the systems studied were performed using the Coulomb attenuated CAM-B3LYP functional,45 which was previously successfully used for the study of excited states.46,47 TDDFT calculations were performed using the DALTON software package.48,49 For all polymer/PCBM combinations the first 10 singlet excitations were calculated. For these the Λ parameter and the oscillator strength (f) were then calculated to get insight in the character of the excitation. The Λ parameter gives a measure of the spatial overlap between the occupied and virtual orbitals involved in an excitation, and it was shown that it can be used to gain insight into the observed excitation energy errors for a given exchange–correlation functional. It is given by the following expression:

In this expression, κia represents the contribution of a given occupied-virtual orbital pair (φi(r) and φa(r)) to the excitation; Oia is a measure of the spatial overlap between these orbitals which is computed as  . For more details, we refer to ref. 44. The oscillator strengths obtained from TDDFT have been shown to agree with experimental results when hybrid functionals are used, but it was noted that the amount of exact exchange has a significant influence on the f values calculated.50 While the use of larger basis sets as well as diffuse functions is advised in order to improve the accuracy of f values,50 the size of the systems involved in this work made this approach unrealistic, and the values obtained for f should be used in the frame of relative trends. In order to reduce the system size, the molecular structure of all donor polymers except P3HT was simplified by replacing aliphatic chains that do not contribute to the π-conjugated backbone by methyl groups (simplified structures can be found in Fig. 1). It has been proven that these alkyl chains do not affect the electronic properties.51 These simplified structures were optimized separately and afterwards in combination with an unsimplified PCBM molecule. PCBM was left unsimplified as it was seen that the excitation scheme of a combined polymer/C60 system differed significantly from that of the corresponding polymer/PCBM system.
. For more details, we refer to ref. 44. The oscillator strengths obtained from TDDFT have been shown to agree with experimental results when hybrid functionals are used, but it was noted that the amount of exact exchange has a significant influence on the f values calculated.50 While the use of larger basis sets as well as diffuse functions is advised in order to improve the accuracy of f values,50 the size of the systems involved in this work made this approach unrealistic, and the values obtained for f should be used in the frame of relative trends. In order to reduce the system size, the molecular structure of all donor polymers except P3HT was simplified by replacing aliphatic chains that do not contribute to the π-conjugated backbone by methyl groups (simplified structures can be found in Fig. 1). It has been proven that these alkyl chains do not affect the electronic properties.51 These simplified structures were optimized separately and afterwards in combination with an unsimplified PCBM molecule. PCBM was left unsimplified as it was seen that the excitation scheme of a combined polymer/C60 system differed significantly from that of the corresponding polymer/PCBM system.
 | ||
| Fig. 1 Simplified structures of the polymers studied with their full unsimplified systematic names. | ||
Results and discussion
Chain length
Geometry optimized chains of up to 8 repeating units were investigated with TDDFT calculations for the six structures under study. The first excitation energy, corresponding to a HOMO → LUMO transition, was investigated in function of chain length to determine the chain lengths used for the remainder of this study. As can be seen in Fig. 2, a chain length of 6 repeating units is in all cases sufficient to reach a stable excitation energy. This corresponds with findings from McCormick et al., where it was noted that for simple conjugated polymers 6 repeating units were sufficient for frontier orbital stability; this dropped to about 4 repeating units for donor–acceptor copolymers.52 However, for the two largest structures (PCDTBT and APFO3), this would lead to an unmanageable system size when PCBM is taken into account. Therefore three repeating unit chains will be used in this case. A length of 3.9 Angstrom was calculated for the thiophene units in a 6 unit P3HT chain, corresponding to experimental results53,54 as well as previous theoretical investigations.23 | ||
| Fig. 2 HOMO → LUMO excitation energy of oligomers in function of their length. | ||
P3HT excitations at the interface with PCBM
Three combinations of P3HT/PCBM with a different starting configuration were considered and full geometry optimizations performed (see Fig. 2). As the real system consists of two semi-crystalline components, a large variation in the possible configurations is expected in reality. For the first geometry the P3HT chain is oriented diametrically opposed to the functional group of the PCBM molecule, shown as configuration A in Fig. 3. This optimized configuration corresponds to the one mentioned by Marchiori and Koehler, where the distance between polymer chain and fullerene is around 3.5 Å.23 The other two starting configurations lead to either a configuration were the chain runs parallel to the PCBM functional group (B configuration), or where the functional groups on the PCBM act as a spacer, shielding the C60 from the P3HT chain (C configuration). The A, B and C configurations found are very similar to some of the configurations calculated by Liu et al., where a combination of molecular dynamics and DFT was used.18 In a more recent study by Marchiori and Koehler, P3HT and PCBM were geometry optimized separately, and the influence of the angle between the two on the energy of the system was investigated.55 Two of the minima found in this study correspond to the A and B configuration described above. In light of these literature results, the three configurations calculated here can be considered reliable structures. | ||
| Fig. 3 Fully optimized geometries results for the three P3HT/PCBM systems considered. Hydrogen atoms are not shown for clarity. | ||
The electronic properties of these three configurations were investigated by TDDFT calculations, and compared to those of the same P3HT chain without the presence of PCBM. A first effect of introducing the PCBM acceptor molecule is that energetically five molecular orbitals localized on the C60 part of the PCBM molecule are found in between the HOMO of the combined system and the first virtual orbital localized on the P3HT chain (LUMO+5), which corresponds to the LUMO of the P3HT chain without PCBM. For the C configuration only three of the C60 molecular orbitals are found in the energetic range mentioned above. It seems that this orientation of the PCBM molecule induces a change in the energetic ordering of the molecular orbitals. It should be noted that this ordering is also achieved for the A and B configurations when periodic boundary conditions (PBC) are used in combination with a unit cell of 6 P3HT repeating units and one PCBM molecule. We therefore conjecture that the three PCBM molecular orbitals that lie between the HOMO of the combined system and the first virtual orbital localized on the P3HT chain are to be considered for charge transfer, this is confirmed by TDDFT results below. The use of PBC however was unpractical in combination with a TDDFT excitation study due to the system sizes involved. The final geometries of the three configurations discussed can be found in the ESI.†
From TDDFT calculations on the interacting polymer/PCBM system, the only singlet transition with a significant value for f found in the A and B configurations corresponds to a HOMO → LUMO+5 transition (respectively S9 and S10, both with an excitation energy of 2.93 eV), while for the C conformation the situation is more complex with a significant f transition corresponding to a HOMO → LUMO+3 transition and a second significant f transition involving deeper lying orbitals. However, due to the difference in energetical ordering for the C conformation the LUMO+3 molecular orbital can be identified with the LUMO+5 seen in the A and B conformation. When these results are compared to the excitation scheme for only P3HT, the similar transition S1,P3HT is also the only one with significant f (2.07) and it corresponds to the HOMO → LUMO transition. It can thus be concluded that the only important transition for the combined systems is the photo-excitation of the polymer chain. These transitions have a high Λ value, indicating that there is a high spatial overlap between the orbitals involved.
Table 1 summarizes the 10 singlet excitations calculated for the A configuration. Direct excitations, from the P3HT polymer to the PCBM donor, are present with low f values for the two configurations where the C60 group faces the polymer chain, e.g. S1–S3 for the A configuration. These transitions also exhibit a low Λ value, giving further proof that they involve mostly molecular orbitals localized on the two different molecules, and that these excitations belong to the class of charge transfer excitations. Several local, high Λ dark excitations also occur, which can be attributed mainly to molecular orbitals localized on the P3HT chain. Visualizations of the molecular orbitals involved in S9 (the excitation of the polymer) and S1 (the HOMO → LUMO charge transfer) can be seen in Fig. 4. The extra virtual orbitals LUMO+3 and LUMO+4 mentioned before, whose ordering changes when PBC are used, play no significant role in the first 10 singlet excitations, and as such are not expected to play a role in charge transfer. Experimentally a maximum in the absorption spectrum is found at 485 nm for a P3HT/PCBM active layer that has not undergone any thermal treatment.56 This corresponds to 2.56 eV, meaning that the value calculated for the S9 transition of the A conformation is overestimated by 0.37 eV. As experimental values are obtained for solid films, where the absorption maximum depends on the degree of crystallinity,56 a deviation with gas-phase calculations is to be expected. Even in this case such a deviation is within the maximum errors seen by Peach et al. for a test set of selected molecules.46
| Excitation | Excitation energy (eV) | Λ | f |
|---|---|---|---|
| S1,P3HT | 2.86 | 0.80 | 2.07 |
| S1 | 2.15 | 0.11 | 8.06 × 10−4 |
| S2 | 2.21 | 0.15 | 4.75 × 10−2 |
| S3 | 2.40 | 0.12 | 7.09 × 10−3 |
| S4 | 2.62 | 0.62 | 5.15 × 10−3 |
| S5 | 2.67 | 0.74 | 1.53 × 10−4 |
| S6 | 2.74 | 0.58 | 3.19 × 10−6 |
| S7 | 2.77 | 0.70 | 5.24 × 10−5 |
| S8 | 2.91 | 0.54 | 5.27 × 10−4 |
| S9 | 2.93 | 0.74 | 1.72 |
| S10 | 2.94 | 0.65 | 1.31 × 10−1 |
 | ||
| Fig. 4 Visualizations of the dominant molecular orbitals involved in the high f S9 excitation of the donor polymer (top) and the S1 dark excitation corresponding to charge transfer (bottom). The A configuration is shown here. Hydrogen atoms are not shown for clarity. | ||
When the triplet excitations are examined, the T1 excitation seems to correspond to the S9 HOMO → LUMO+5 transition, or the excitation of the polymer, which was the significant singlet transition. At an excitation energy of 1.50 eV it would be unlikely that this transition is of direct importance, as this would lie in the infra-red region of the solar spectrum. Furthermore, this transition could not lead to charge generation according to the results in this work, as the direct transitions exhibit a higher excitation energy. This would indicate that the T1 is a lower lying state compared to the singlet excited states involving virtual orbitals localized on the PCBM molecule (S1, S2, S3), and a charge transfer would not lead to a stabilization. However, this may be a possible loss mechanism, because electrons in this triplet state can not contribute to charge generation. This is in correspondence with the remarks made by Brédas et al. regarding triplet states.57 Furthermore it is possible for the triplet state to play a role in recombination.
It should be noted however that in work by Peach et al., it was seen that functionals incorporating exact exchange, such as CAM-B3LYP, can lead to triplet instability errors, leading to an underestimation of the triplet excitation energies.58 A possible solution for these errors would be the application of the Tamm–Dancoff approximation (TDA). However, as the triplet excitations are not a primary concern of this work, only singlet excitations will be taken into account. A scheme with the most important excitations and their ordering with respect to the ground state of the interacting P3HT/PCBM system can be seen in Fig. 5 for the A configuration.
 | ||
| Fig. 5 Scheme of the most important excitations for the interacting donor–acceptor A configuration (see Fig. 3) system and their ordering with respect to the ground state. | ||
The C configuration where the functional groups of the PCBM molecule act as a spacer has less of the low f and Λ charge transfer excitations. This can be explained by the much larger distance between the polymer and C60 orbitals. Combined with the different excitation scheme mentioned above, it can be concluded that for this configuration the polymer chain and PCBM molecule interact less due to the increased distance between the polymer chain and the C60 group of the PCBM molecule. No evidence is found of any other excitation mechanism, such as photo-excitation of the PCBM molecule. As the excitation scheme for the A and B configurations is very similar, it can be concluded that the exact position of the PCBM acceptor does not have a major effect on the electronic properties, as long as it is oriented towards the donor polymer chain, and the distance between donor and acceptor is thus comparable. Therefore only one optimized configuration satisfying this condition will be discussed for the other donor polymers under study, as an in-depth study of the effects of the geometry on the electronic properties does not fit in the frame of this work. Using similar optimized donor–acceptor conformations provides a more consistent method to investigate the intrinsic electronic properties of the different systems under study, as the geometry effects are excluded.
Excitation behavior of the other polymers at the interface with PCBM
Systems consisting of interacting polymer chain segments and the PCBM molecule were optimized is a similar way as for P3HT/PCBM, the final geometry files are provided as ESI.† In analogy with the previous section, TDDFT was used to study the excitation behaviour of these systems. A summary of the excitations calculated for these systems can be found in Table 2. Surprisingly, the excitation schemes for the other donor polymer/PCBM combinations look similar qualitatively, with one important (high f) transition corresponding to the excitation of the donor polymer, which can be abbreviated as D → D*, and can be linked to a transition from the HOMO to the first virtual orbital localized on the polymer chain. This transition always shows a high Λ value, further proving the local character. No other excitations with high f are found among the first 10 singlet excitations. The other low intensity excitations can be divided in those with a high and a low value for the Λ parameter. The former group comprises the local excitations between molecular orbitals localized primarily on the polymer chain, while the latter are the charge transfer excitations between polymer chain and PCBM molecule. A notable exception is the PBDTTPD system, where an extra high f transition is seen, corresponding both to a transition from the HOMO−1 to the first virtual orbital localized on the polymer, and from the HOMO to the second virtual orbital localized on the polymer. Experimentally, a double maximum can also be seen in the absorption spectrum, confirming these results.29 A possible explanation is the highly planar nature of PBDTTPD, also seen in this work, and which promotes delocalization effects.59 The orbital contributions of the excitations found in Table 2 can be found in the ESI.† When the large f excitations are compared to the absorption maxima of experimental spectra, obtained from solid films (and dissolved in toluene for MDMO-PPV/PCBM),21,29,38,60,61 it is again seen that the excitation energies calculated are overestimated compared to experimental values. As was discussed above, this can be expected from gas-phase calculations. The largest deviation is found for the S1 excitation of PBDTTPD, with an experimental value of about 630 nm or 1.97 eV compared to a calculated value of 2.56 eV. This is still below the maximum positive deviation seen by Peach et al. for a test set of selected molecules.46| PCDTBT/PCBM | |||
|---|---|---|---|
| Excitation | Excitation energy (eV) | Λ | f |
| S1,PCDTBT | 2.47 | 0.54 | 2.59 |
| S1 | 2.49 | 0.60 | 3.21 |
| S2 | 2.59 | 0.59 | 2.20 × 10−1 |
| S3 | 2.63 | 0.55 | 2.40 × 10−3 |
| S4 | 2.67 | 0.68 | 3.69 × 10−5 |
| S5 | 2.72 | 0.14 | 1.57 × 10−1 |
| S6 | 2.74 | 0.50 | 3.30 × 10−1 |
| S7 | 2.74 | 0.08 | 1.02 × 10−2 |
| S8 | 2.76 | 0.65 | 1.89 × 10−4 |
| S9 | 2.77 | 0.71 | 8.26 × 10−6 |
| S10 | 2.90 | 0.08 | 1.76 × 10−3 |
| PCPDTBT/PCBM | |||
|---|---|---|---|
| Excitation | Excitation energy (eV) | Λ | f |
| S1,PCPDTBT | 1.85 | 0.68 | 6.49 |
| S1 | 1.86 | 0.60 | 6.30 |
| S2 | 2.05 | 0.59 | 1.81 × 10−2 |
| S3 | 2.26 | 0.50 | 5.09 × 10−1 |
| S4 | 2.32 | 0.21 | 2.54 × 10−2 |
| S5 | 2.34 | 0.36 | 2.87 × 10−2 |
| S6 | 2.43 | 0.54 | 4.48 × 10−2 |
| S7 | 2.52 | 0.50 | 7.78 × 10−2 |
| S8 | 2.62 | 0.51 | 4.74 × 10−2 |
| S9 | 2.63 | 0.56 | 6.18 × 10−3 |
| S10 | 2.68 | 0.67 | 8.41 × 10−4 |
| APFO3/PCBM | |||
|---|---|---|---|
| Excitation | Excitation energy (eV) | Λ | f |
| S1,APFO3 | 2.45 | 0.54 | 2.56 |
| S1 | 2.48 | 0.55 | 2.83 |
| S2 | 2.59 | 0.53 | 2.82 × 10−1 |
| S3 | 2.62 | 0.60 | 6.96 × 10−3 |
| S4 | 2.64 | 0.13 | 8.93 × 10−2 |
| S5 | 2.67 | 0.73 | 6.86 × 10−3 |
| S6 | 2.70 | 0.18 | 2.48 × 10−1 |
| S7 | 2.74 | 0.53 | 3.89 × 10−1 |
| S8 | 2.76 | 0.61 | 1.37 × 10−4 |
| S9 | 2.76 | 0.73 | 6.69 × 10−5 |
| S10 | 2.85 | 0.26 | 4.71 × 10−2 |
| PBDTTPD/PCBM | |||
|---|---|---|---|
| Excitation | Excitation energy (eV) | Λ | f |
| S1,PBDTTPD | 2.27 | 0.28 | 3.10 × 10−2 |
| S2,PBDTTPD | 2.81 | 0.56 | 5.98 × 10−1 |
| S1 | 2.56 | 0.66 | 3.85 |
| S2 | 2.63 | 0.48 | 7.43 × 10−3 |
| S3 | 2.67 | 0.59 | 7.99 × 10−3 |
| S4 | 2.70 | 0.63 | 2.87 |
| S5 | 2.73 | 0.17 | 8.42 × 10−2 |
| S6 | 2.75 | 0.35 | 3.24 × 10−3 |
| S7 | 2.75 | 0.64 | 4.58 × 10−3 |
| S8 | 2.79 | 0.37 | 6.15 × 10−3 |
| S9 | 2.83 | 0.54 | 3.09 × 10−3 |
| S10 | 2.90 | 0.49 | 8.50 × 10−2 |
| MDMO-PPV/PCBM | |||
|---|---|---|---|
| Excitation | Excitation energy (eV) | Λ | f |
| S1,MDMO-PPV | 2.72 | 0.78 | 4.61 |
| S1 | 2.07 | 0.11 | 9.56 × 10−3 |
| S2 | 2.10 | 0.10 | 3.22 × 10−2 |
| S3 | 2.32 | 0.12 | 2.34 × 10−2 |
| S4 | 2.62 | 0.47 | 7.72 × 10−3 |
| S5 | 2.65 | 0.52 | 1.97 × 10−3 |
| S6 | 2.70 | 0.59 | 4.02 |
| S7 | 2.73 | 0.18 | 2.84 × 10−1 |
| S8 | 2.76 | 0.67 | 2.40 × 10−3 |
| S9 | 2.77 | 0.52 | 2.42 × 10−3 |
| S10 | 2.77 | 0.74 | 6.51 × 10−3 |
Charge transfer
In all donor polymer/PCBM cases low intensity charge transfer excitations are found, that can be attributed to excitations from the polymer localized HOMO of the combined system to one of the three lowest virtual molecular orbitals localized on PCBM.The low intensities indicate, as can be expected, that the direct excitation from donor to acceptor is insignificant. In the generally accepted mechanism of charge transfer, the excited donor polymer transfers an electron to an acceptor molecule. This creates a bound electron–hole pair that will dissociate if the energy difference between the two molecular orbitals involved is large enough. The first phase, before dissociation of the bound pair, is thus a transition between the orbital corresponding to the first virtual orbital located on the polymer chain and one of the three close-lying virtual orbitals on the fullerene of the PCBM molecule (see Fig. 6). Such a transition between two virtual orbitals can not be calculated directly by TDDFT, but two important quantities involved in it can: the intensity fD→D* of the excitation of the polymer (as in the previous section), and the orbital overlaps Oia between donor and acceptor molecular orbitals involved, which is used for the calculation of the Λ parameter. When the Oia values are calculated an immediate observation is the rather small three values for the PCDTBT/PCBM system, the highest Oia found is only about 0.05 while for the other systems at least one value is above 0.1. For PCPDTBT on the other hand, a different energetical order is observed, where the first virtual molecular orbital localized on the polymer chain is found in between the second and third virtual orbital on PCBM. Because of this, only the Oia values between the first virtual orbital on the polymer chain and the two lower virtual orbitals are taken into account.
 | ||
| Fig. 6 Visualizations of the three lowest virtual orbitals of the PCBM molecule, these also correspond to the lowest virtual orbitals of the combined systems. | ||
When all the Oia are brought together, a trend is seen where the more recently developed donor–acceptor copolymers have a lower average overlap than the two conventional polymers (values can be found in Table 3). The commercial PCDTBT, which has shown the highest efficiencies of the polymers tested, clearly shows the lowest average Oia, followed by APFO 3, PBDTTPD, PCPDTBT, P3HT and MDMO-PPV. The average Oia of PCPDTBT seems to be quite high when compared to the other similar polymers, which can be explained by only taking two overlaps into account instead of three. If only the Oia with the first virtual orbital on PCBM is taken into account, PCPDTBT actually shows the second lowest value. Visualizations of all the molecular orbitals taken into consideration for this methodology can be found in the ESI.† While the efficiency of an organic solar cell depends on many parameters, including the electronic properties of both components, light absorption, the morphology of the separate phases, the contacts with the electrodes, etc. it can be assumed that the record efficiencies found in literature have been found for cells where most of these parameters are optimized (see the Introduction). It should be noted that the value for P3HT/PCBM is chosen to represent a simple P3HT/PCBM active layer, without extra components, as these are also not taken into account in the study. In this case the relative value of the efficiencies may give a clue as to how successful the charge transfer is.
| P3HT | MDMO-PPV | APFO3 | PBDTTPD | PCDTBT | PCPDTBT | |
|---|---|---|---|---|---|---|
| Oia LUMO+2 | 0.21 | 0.40 | 0.28 | 0.24 | 0.05 | — |
| Oia LUMO+1 | 0.22 | 0.40 | 0.13 | 0.15 | 0.05 | 0.33 |
| Oia LUMO | 0.21 | 0.40 | 0.10 | 0.12 | 0.03 | 0.08 |
| Average Oia | 0.21 | 0.40 | 0.17 | 0.17 | 0.04 | 0.21 |
| fD→D* | 1.72 | 4.02 | 2.83 | 3.85 | 3.21 | 6.30 |
| fD→D*/average Oia | 8.19 | 10.05 | 16.65 | 22.65 | 80.25 | 30.00 |
| Efficiencies (literature) | 5.0 (ref. 30) | 2.9 (ref. 32) | 3.5 (ref. 36) | 6.8 (ref. 37) | 7.5 (ref. 33) | 5.5 (ref. 35) |
A possible explanation of a higher efficiency when the overlap is actually lower may then lie in the lower possibility of recombination. Where the actual charge transfer event would then require a low Oia, recombination may actually be more successful when the overlap of the molecular orbitals involved is higher. This seems logical when the overlap between molecular orbitals is considered as a measure of the spatial distance between the electron and the hole, on the acceptor and donor respectively. The order seen from Oia values seems to show similarities with the efficiencies seen in literature. When the oscillator strength fD→D* is also taken into account by dividing this value with the average Oia, it can be seen that this value correlates roughly with the efficiency (see Fig. 7).
 | ||
| Fig. 7 The fD→D*/average Oia quotient in function of record efficiencies from literature for the systems under study. As a guide to the eye a trend line is also shown. | ||
The difficulty of discerning such a trend can be illustrated by taking into account several efficiencies found in literature instead of just the record efficiencies.29,33,61–65 This leads to efficiency-intervals with important overlaps as can be seen in Fig. 8, further demonstrating the complex nature of the retrieved experimental efficiencies, depending on many experimental parameters as well as intrinsic material properties.
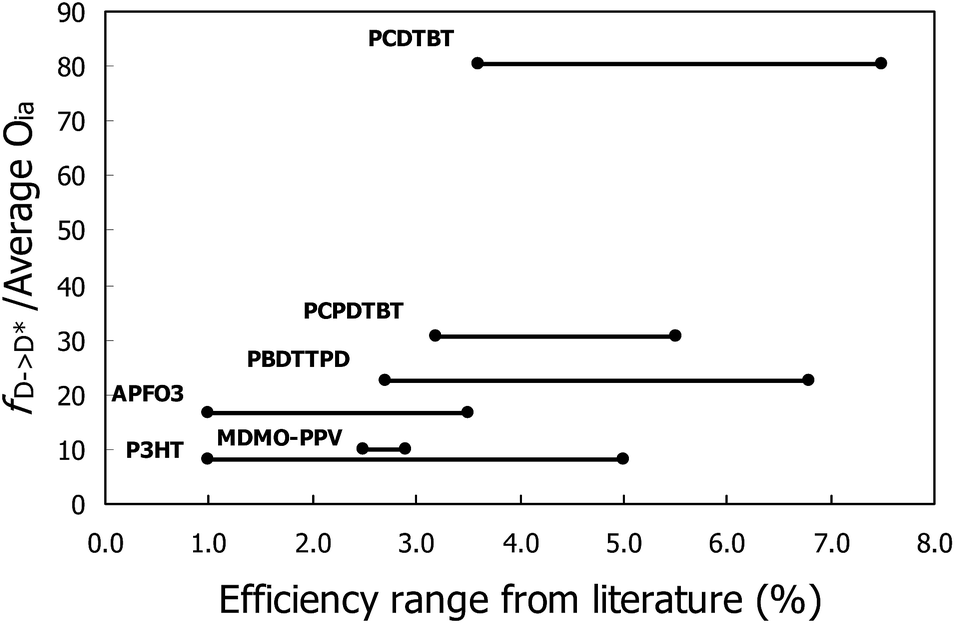 | ||
| Fig. 8 The fD→D*/average Oia quotient plotted for efficiency intervals that summarize the extent of efficiencies found in literature for each polymer/PCBM combination. | ||
Conclusion
In this work, the singlet excitations at the interface with a PCBM acceptor were investigated by TDDFT for two conventional and four donor–acceptor polymers used in organic photovoltaic devices. The P3HT/PCBM system was taken as the model system for the other polymer/PCBM combinations. From the results on this model system it could be concluded that no significant differences are seen in the excitation scheme of different geometry optimized P3HT/PCBM configurations as long as the fullerene group of the PCBM molecule is oriented towards the polymer chain. Consequently, a single geometry optimized configuration was used for the excitation study of the other polymer/PCBM combinations.A general conclusion that could be made for all systems is that the only intense excitation corresponded to the excitation of the donor polymer. These excitations also show a high Λ parameter value, further stressing the local character of these excitations. When the excitations of the combined systems are compared with those of the pure polymers, differences can be seen in the excitation energies and oscillator strength f of the polymer excitation. This indicates the important difference between the electronic properties of the donor polymer at the interface and in bulk. Furthermore, the appearance of several dark transitions with low intensity and Λ parameter was seen. These can be identified as charge transfer excitations between donor polymer and PCBM acceptor. In an attempt to link the efficiency of charge transfer to an easily accessible parameter from TDDFT, efficiencies from literature were compared to the orbital overlaps Oia between the molecular orbitals involved in charge transfer and the excitation strength f of the polymer excitation. A broad correlation could be discerned, with more efficient donor–PCBM pairs demonstrating a lower average overlap.
Acknowledgements
The authors would like to thank Dr M.J.G. Peach (Lancaster University) and Professor D. J. Tozer (Durham University) for many fruitful discussions on the topic of TDDFT and the Λ parameter. The authors acknowledge support from the Research Foundation Flanders (FWO); the Vrije Universiteit Brussel (VUB) through a Strategic Research Program for the ALGC group; and the COST Materials, Physical and Nanosciences (MPNS) Action MP0901: “ Designing Novel Materials for Nanodevices: From Theory to Practice (NanoTP)”.Notes and references
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- B. A. Gregg, J. Phys. Chem. B, 2003, 107, 4688–4698 CrossRef CAS.
- L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642–6671 CrossRef CAS PubMed.
- M. T. Dang, L. Hirsch, G. Wantz and J. D. Wuest, Chem. Rev., 2013, 113, 3734–3765 CrossRef CAS PubMed.
- D. Venkataraman, S. Yurt, B. H. Venkatraman and N. Gavvalapalli, J. Phys. Chem. Lett., 2010, 1, 947–958 CrossRef CAS.
- C. J. Brabec, S. Gowrisanker, J. J. M. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839–3856 CrossRef CAS PubMed.
- A. L. Ayzner, C. J. Tassone, S. H. Tolbert and B. J. Schwartz, J. Phys. Chem. C, 2009, 113, 20050–20060 CAS.
- D. Yu, Y. Yang, M. Durstock, J. Baek and L. Dai, ACS Nano, 2010, 4, 5633–5640 CrossRef CAS PubMed.
- M. Hilczer and M. Tachiya, J. Phys. Chem. C, 2010, 114, 6808–6813 CAS.
- S. K. Pal, T. Kesti, M. Maiti, F. Zhang, O. Inganäs, S. Hellström, M. R. Andersson, F. Oswald, F. Langa, T. Osterman, T. Pascher, A. Yartsev and V. Sundström, J. Am. Chem. Soc., 2010, 132, 12440–12451 CrossRef CAS PubMed.
- P. M. Beaujuge, H. N. Tsao, M. R. Hansen, C. M. Amb, C. Risko, J. Subbiah, K. R. Choudhury, A. Mavrinskiy, W. Pisula, J. Bre, F. So, K. Mu and J. R. Reynolds, J. Am. Chem. Soc., 2012, 134, 8944–8957 CrossRef CAS PubMed.
- H. Van Mullekom and J. Vekemans, Mater. Sci. Eng., R, 2001, 32, 1–40 CrossRef.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- F. Zhang, Z. Zhuo, J. Zhang, X. Wang, X. Xu, Z. Wang, Y. Xin, J. Wang, J. Wang, W. Tang, Z. Xu and Y. Wang, Sol. Energy Mater. Sol. Cells, 2012, 97, 71–77 CrossRef CAS PubMed.
- N. Van den Brande, F. Demir, P. Geerlings, B. Van Mele, G. Van Lier, and G. Van Assche, in Proceedings of SPIE, ed. B. P. Rand, C. Adachi, and V. van Elsbergen, 2012, vol. 8435, p. 84352E Search PubMed.
- J. Nelson, J. J. Kwiatkowski, J. Kirkpatrick and J. M. Frost, Acc. Chem. Res., 2009, 42, 1768–1778 CrossRef CAS PubMed.
- T. Liu, D. L. Cheung and A. Troisi, Phys. Chem. Chem. Phys., 2011, 13, 21461–21470 RSC.
- A. Azazi, A. Mabrouk and K. Alimi, Comput. Theor. Chem., 2011, 978, 7–15 CrossRef CAS PubMed.
- X. Lu, C. Wu, S. Wei and W. Guo, J. Phys. Chem. A, 2009, 114, 1178–1184 CrossRef PubMed.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed.
- L. Zhang, K. Pei, H. Zhao, S. Wu, Y. Wang and J. Gao, Chem. Phys. Lett., 2012, 543, 199–204 CrossRef CAS PubMed.
- C. F. N. Marchiori and M. Koehler, Synth. Met., 2010, 160, 643–650 CrossRef CAS PubMed.
- Y. Kanai and J. C. Grossman, Nano, 2007, 7, 2–7 Search PubMed.
- J. I. Fuks, A. Rubio and N. T. Maitra, Phys. Rev. A: At., Mol., Opt. Phys., 2011, 83, 042501 CrossRef.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed.
- M. J. G. Peach, C. R. Le Sueur, K. Ruud, M. Guillaume and D. J. Tozer, Phys. Chem. Chem. Phys., 2009, 11, 4465–4470 RSC.
- O. Inganäs, F. Zhang and M. R. Andersson, Acc. Chem. Res., 2009, 42, 1731–1739 CrossRef PubMed.
- Y. Zou, A. Najari, P. Berrouard and S. Beaupre, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS PubMed.
- W. Ma, C. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- C.-Y. Chang, C.-E. Wu, S.-Y. Chen, C. Cui, Y.-J. Cheng, C.-S. Hsu, Y.-L. Wang and Y. Li, Angew. Chem., Int. Ed., 2011, 50, 9386–9390 CrossRef CAS PubMed.
- T. Munters, T. Martens, L. Goris, V. Vrindts, J. Manca, L. Lutsen, W. De Ceuninck, D. Vanderzande, L. De Schepper, J. Gelan, N. S. Sariciftci and C. J. Brabec, Thin Solid Films, 2002, 403, 247–251 CrossRef.
- S. Beaupré and M. Leclerc, J. Mater. Chem. A, 2013, 1, 11097–11105 Search PubMed.
- J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619–3623 CrossRef CAS PubMed.
- J. Peet, J. Kim, N. Coates, W. Ma, D. Moses, A. Heeger and G. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- C. M. Björnström Svanström, J. Rysz, A. Bernasik, A. Budkowski, F. Zhang, O. Inganäs, M. R. Andersson, K. O. Magnusson, J. J. Benson-Smith, J. Nelson and E. Moons, Adv. Mater., 2009, 21, 4398–4403 CrossRef.
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595–7597 CrossRef CAS PubMed.
- M. M. Wienk, J. M. Kroon, W. J. H. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. J. Janssen, Angew. Chem., Int. Ed., 2003, 42, 3371–3375 CrossRef CAS PubMed.
- S. Cook, R. Katoh and A. Furube, J. Phys. Chem. C, 2009, 113, 2547–2552 CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS PubMed.
- J. S. Binkley, J. A. Pople and W. J. Hehre, J. Am. Chem. Soc., 1980, 102, 939–947 CrossRef CAS.
- M. Niskanen and T. I. Hukka, Phys. Chem. Chem. Phys., 2014, 16, 13294–13305 RSC.
- D. Jacquemin, A. Femenias, H. Chermette, I. Ciofini, C. Adamo, J.-M. André and E. A. Perpète, J. Phys. Chem. A, 2006, 110, 5952–5959 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision B.01, 2009 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS PubMed.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed.
- M. J. G. Peach, E. I. Tellgren, P. Sałek, T. Helgaker and D. J. Tozer, J. Phys. Chem. A, 2007, 111, 11930–11935 CrossRef CAS PubMed.
- Dalton, A Molecular Electronic Structure Program, Release DALTON, 2011, see http://daltonprogram.org.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenaes, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjaergaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnaes, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, The Dalton Quantum Chemistry Program System, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 4, 269–284 CrossRef PubMed.
- M. Miura, Y. Aoki and B. Champagne, J. Chem. Phys., 2007, 127, 084103 CrossRef PubMed.
- M. Belletête, S. Beaupre, J. Bouchard, P. Blondin, M. Leclerc and G. Durocher, J. Phys. Chem. B, 2000, 104, 9118–9125 CrossRef.
- T. M. McCormick, C. R. Bridges, E. I. Carrera, P. M. Dicarmine, G. L. Gibson, J. Hollinger, L. M. Kozycz and D. S. Seferos, Macromolecules, 2013, 46, 3879–3886 CrossRef CAS.
- T. J. Prosa, M. J. Winokur, J. Moulton, P. Smith and A. J. Heeger, Macromolecules, 1992, 25, 4364–4372 CrossRef CAS.
- V. I. Arkhipov, P. Heremans and H. Bässler, Appl. Phys. Lett., 2003, 82, 4605–4607 CrossRef CAS PubMed.
- C. F. N. Marchiori and M. Koehler, J. Phys. D: Appl. Phys., 2014, 47, 215104 CrossRef.
- T. M. Clarke, F. C. Jamieson and J. R. Durrant, J. Phys. Chem. C, 2009, 113, 20934–20941 CAS.
- J.-L. Brédas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691–1699 CrossRef PubMed.
- M. J. G. Peach and D. J. Tozer, J. Phys. Chem. A, 2012, 116, 9783–9789 CrossRef CAS PubMed.
- I. Hwang, S. Beaupré, M. Leclerc and G. D. Scholes, Chem. Sci., 2012, 3, 2270–2277 RSC.
- S. K. Pal, T. Kesti, M. Maiti, F. Zhang, S. Hellstro, M. R. Andersson, F. Oswald, F. Langa and O. Tomas, J. Am. Chem. Soc., 2010, 12440–12451 CrossRef CAS PubMed.
- D. Mühlbacher, M. Scharber, M. Morana, Z. Zhu, D. Waller, R. Gaudiana and C. Brabec, Adv. Mater., 2006, 18, 2884–2889 CrossRef.
- Organic Photovoltaics Materials, Device Physics, and Manufacturing Technologies, ed. C. Brabec, V. Dyakonov and U. Scherf, WILEY-VCH Verlag, Weinheim, 2008 Search PubMed.
- F. Zhang, K. G. Jespersen, C. Björström, M. Svensson, M. R. Andersson, V. Sundström, K. Magnusson, E. Moons, A. Yartsev and O. Inganäs, Adv. Funct. Mater., 2006, 16, 667–674 CrossRef CAS.
- M. Svensson, F. Zhang, O. Inganäs and M. Andersson, Synth. Met., 2003, 134, 137–138 CrossRef.
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Coordinates of the geometry optimized systems used for the charge transfer study, visualizations of the most important molecular orbitals for all the systems studied and more detailed excitation tables mentioning the molecular orbital contributions. See DOI: 10.1039/c4ra12053a |
| ‡ Present address: Laboratoire des Solides Irradiés, UMR 7642, CNRS-CEA/DSM, École Polytechnique, F-91128 Palaiseau, France and European Theoretical Spectroscopy Facility (ETSF). |
| This journal is © The Royal Society of Chemistry 2014 |