DOI:
10.1039/C4RA12033G
(Paper)
RSC Adv., 2014,
4, 61877-61883
A template induced method to synthesize nanoporous graphitic carbon nitride with enhanced photocatalytic activity under visible light†
Received
9th October 2014
, Accepted 7th November 2014
First published on 11th November 2014
Abstract
A template induced method was developed to synthesize nanoporous graphitic carbon nitride (npg-CN) by directly heating Triton X-100-modified-melamine sulfate. The effects of the added amount of Triton X-100 and heating temperature on the formation of npg-CN were investigated. The npg-CN samples were characterized by XRD, FT-IR, and elemental analysis; nitrogen adsorption isotherms, SEM and TEM images; and UV-vis DRS and photoluminescence spectra. The results showed that the npg-CN samples possessed a laver-like layered structure with a high surface area (50–135 m2 g−1). Meanwhile, compared to the bulk g-CN (synthesized by directly heating melamine), the npg-CN samples possessed stronger optical absorptions, narrower band gaps, and significantly reduced fluorescence intensity. The photocatalytic activity was investigated by degradation of Rhodamine B under visible light irradiation. As a result, the photodegradation rate of Rhodamine B dye on the npg-CN samples was greatly improved over that on bulk g-CN.
1. Introduction
Recently, graphitic carbon nitride (g-CN) has attracted considerable attention because of its catalytic and photocatalytic activity as a new metal-free catalyst.1–3 g-CN materials have been found useful for many applications: as metal-free photocatalyst,4,5 in water splitting,6–8 in pollutant degradation,9,10 and more. Its unique optical, chemical, and catalytic properties, along with its low price and remarkably high stability against oxidation, make it a very attractive material for photocatalytic applications. However, the photocatalytic activities of pristine g-CN are seriously limited by its intrinsic drawbacks, including relatively low surface area, fast charge recombination, and poor mass diffusion/transfer. Many modifications of g-CN have been developed, including texture modification,11,12 nonmetal doping,13–15 and noble metal deposition;5,16 and constructing heterojunctions with other materials.17–19 Among these methods, the texture modification have been demonstrated a successful and efficient pathway to promote charge migration and separation,11 as well as the mass diffusion/transfer during photoredox reactions,12 greatly enhancing the photocatalytic performance. Hard templating strategy has been widely reported and successfully used in the synthesis of nanoporous g-CN (npg-CN) with desired surface properties and morphologies.12,20 But hard templating methods are step tedious and environmentally harmful because the synthesis procedure involves using silica or alumina as hard templates, which need to be subsequently removed by aqueous ammonium bifluoride (NH4HF2), hydrogen fluoride (HF) or NaOH solutions. Therefore, a novel procedure for the synthesis of npg-CN without hard templates is of particular interest. Wang and co-workers21 first tried to explore a soft-template method to synthesize npg-CN by using a variety of surfactants as structure-directing agents and dicyandiamide as precursor. Most experiments yielded disordered pore systems and high molar ratio of C/N (0.8–2.0) because of the carbon residues generated from the decomposition of soft templates. Yan22 used melamine (MA) and P123 to synthesize npg-CN, and obtained the npg-CN samples with low molar ratios of C/N (0.65–0.69) as well as low BET surface areas (about 30 m2 g−1) without further calcination in air. In general, the npg-CN often possesses low surface areas and high molar ratios of C/N because of the surfactant self-decomposition and the side-reaction of the surfactant with the precursor during the thermal treatment. To the best of our knowledge, there are few reports trying to improve soft-template methods of npg-CN synthesis by solving the problems mentioned above.
Herein, a template induced method derived from the soft-template method was proposed to synthesize npg-CN, by which the side-reactions of surfactants can be avoided completely. Because the nonionic surfactant was just used to modify the structure of precursor and then totally removed before the thermal treatment. Melamine sulfate was chosen as the precursor because the sublimation of melamine can be significantly suppressed by treating melamine with sulfuric acid23 and the insolubility of melamine sulfate make it can be easily separated from the nonionic surfactant. Triton X-100 was used to modify the structure of melamine sulfate as it can be easily removed by simple washing. As a result, the as-prepared npg-CN samples possessed high BET surface areas and low molar ratios of C/N. The final samples were characterized in terms of chemical structure, morphology, and optical properties. The photocatalytic activity of the obtained samples was tested by evaluating the degradation of Rhodamine B (RhB) dye under visible light illumination.
2. Experimental section
2.1 Synthesis of Triton X-100-modified-melamine sulfate
All reagents were of analytical grade without further treatment. In a typical synthetic procedure, 5.0 g melamine and a certain amount of Triton X-100 (with mass ratios to melamine of 0.3, 0.5, 0.7 and 1.0) was added into 100 mL distilled water, and then the mixture was heated at 100 °C in an oil bath with stir for 1 hour under refluxing. Then 2 mL concentrated sulfuric acid (98 wt%) was added to the solution dropwise along with white precipitates gradually formed, and the mixture was stirred at 100 °C for another hour. After naturally cooling down to room temperature, the precipitate was filtrated and washed three times with distilled water, and then dried in an oven at 80 °C overnight. The obtained samples were the Triton X-100-modified-melamine sulfate, denoted as MST-0.3, 0.5, 0.7 and 1.0, respectively. For comparison, melamine sulfate (MAS) was also prepared by the same method without adding Triton X-100.
2.2 Synthesis of nanoporous graphitic carbon nitride
5.0 g MST was put into an alumina crucible with a cover and then heated to 500 °C in a muffle furnace for 2 hours in air with a heating rate of 2 °C min−1. Further heat treatment was set at 520, 550, 580 and 600 °C under the same condition for another 2 hours, respectively. The final products were denoted as npg-CN-xy, in which x represents the mass ratios (0.3, 0.5, 0.7, and 1.0) of Triton X-100 to melamine, and y = a, b, c and d, represents the final heating temperature of 520, 550, 580 and 600 °C, respectively. For comparison, the melamine and melamine sulfate were treated with the same heating program, while the further heat treatment was just kept at 550 °C for 2 hours. The obtained samples were denoted as bulk g-CN and CN–MAS, respectively.
2.3 Characterization
X-ray diffraction (XRD) patterns were collected in a Bruker D8FOCUS powder diffractometer with Cu Kα irradiation (λ = 0.15406 nm). The Fourier transform infrared spectra (FTIR) of the samples were performed using a Nicolet iS10 spectrometer. The scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images were taken with Hitachi S4700 and Tecnai G220 microscope, respectively. Nitrogen sorption measurements were accomplished with N2 at 77 K after degassing the samples at 300 °C under vacuum for 3 hours using a Quantachrome Quadrasorb SI-MP porosimeter. The specific surface area was calculated by applying the Brunauer–Emmett–Teller (BET) model to the adsorption data. Elemental analyses (C, H, N, S) were performed on a EuroVector S.P.A. Euro EA3000 elemental analyzer. UV-vis diffuse reflectance spectra (UV-vis DRS) were measured using a Shimadzu UV3600 spectrophotometer. The photoluminescence (PL) spectra were performed on a Hitachi F-7000 fluorescence spectrophotometer with photomultiplier tube voltage of 400 V and scanning speed of 1200 nm min−1. The TG-DTA curves were recorded on DTG-60A (Shimadzu) at a heating rate of 10 K min−1 under the flow of N2 gas.
2.4 Photocatalytic evaluation
The photocatalytic activity of the samples was evaluated by degradation of RhB under visible light irradiation. A 500 W Xenon lamp (Institute of Electric Light Source, Beijing) with 400 nm cutoff filter was chosen as a visible light source. The temperature of the reaction solution was kept at approximately 20 °C by equipped with a cooling water sleeve surrounding the lamp. The distance between the light source and the surface of the reaction solution was 15 cm. In a typical experiment, 0.1 g of photocatalyst was suspended in RhB aqueous solution (100 mL, 10 mg L−1), and then stirred in the dark for 1 hour to reach the adsorption–desorption equilibrium. During the visible light irradiation, an aliquot of 5 mL containing the sample was taken from the reaction suspensions at given time intervals, and then centrifuged to remove the photocatalyst particles. Subsequently, the solutions were measured with the UV-vis spectrophotometer at wavelength of 554 nm. The concentration changes were described by C/C0, where C0 is the initial concentration of RhB (after subtraction of the concentration of RhB which was adsorbed to the catalysts) and C is the remained concentration of RhB. The RhB degradation ratio (DR) was determined by DR = [1 − (C/C0)] × 100%. The degradation rate was monitored by the rate constant (k), which can be calculated from the fitted curve of the first-order equation: ln(C0/C) = kt.
3. Results and discussion
3.1 Chemical structure and texture of npg-CN samples
The chemical structure of the npg-CN samples was characterized by means of XRD, FT-IR, and elemental analysis. Fig. 1 shows the XRD patterns and FT-IR spectra of npg-CN samples prepared with different mass ratios of Triton X-100 to melamine at 550 °C. The XRD patterns (Fig. 1a) of all the npg-CN samples are similar to that of bulk g-CN. The strongest peak around 2θ = 27.4° is attributed to the typical graphitic interlayer (002) peak with d = 0.326 nm, and another peak at about 2θ = 13°, corresponding to a distance of 0.67 nm and indexed as (100) spacing of g-CN, can be attributed to the in-plane structural packing motif of g-CN.24 Compared with that of bulk g-CN, the (002) peaks of npg-CN samples shift to slightly lower angle and becomes broader with reducing intensity, indicating the increased interlayer distance and the less crystallinity of npg-CN samples. But the higher heating temperature results in higher crystallinity of npg-CN (Fig. S1†), corresponding to previous reports.23,25 The FT-IR (Fig. 1b) spectra show that all samples exhibit several major bands centered at about 3180, 2185, 1200–1700 and 803 cm−1. The broad 3180 cm−1 band can be attributed to the stretching vibration of N–H groups. All samples show stretching modes in the 1200–1700 cm−1 range, which are typical for aromatic C–N heterocycles and originate from the extended CN network. In addition, the characteristic breathing mode of the triazine unit is observed at 803 cm−1.26 The FT-IR spectra of the samples prepared at different temperatures also show the same IR bands (Fig. S2†). Elemental analysis results (Table 1) show the atomic C/N ratio of npg-CN samples are all of same as 0.66, very close to the bulk g-CN (0.65). The lower atomic C/N ratios than the theoretical value (0.75) indicates that the g-CN samples obtained in our experimental conditions are nitrogen-enriched. Besides, all the samples contain H elements indicates that the uncondensed amino groups exist in the samples, corresponding to the FT-IR results. This result demonstrates that there is no carbon residues of Triton X-100 left in the npg-CN samples. The very little S content of npg-CN samples demonstrates that all the sulfate radicals had decomposed.
 |
| | Fig. 1 XRD patterns (a) and FT-IR spectra (b) of bulk g-CN, CN–MAS and npg-CN-(0.3b–1.0b). | |
Table 1 Preparation conditions, physicochemical properties, and photocatalytic activity of bulk g-CN, CN–MAS and npg-CN samples
| Samples |
Product massa (g) |
C/N (atomic) |
H (wt%) |
S (wt%) |
SBETb (m2 g−1) |
Band gap (eV) |
PRc (%) |
DRd (%) |
ke (min−1) |
| 1 g of starting melamine. The BET surface area. The physical adsorption ratio. The RhB degradation ratio under visible light for 90 min. The photodegradation rate constant. |
| Bulk g-CN |
0.46 |
0.65 |
1.94 |
— |
15.1 |
2.58 |
6.0 |
9.0 |
0.00083 |
| CN–MAS |
0.41 |
0.66 |
2.05 |
0.019 |
44.5 |
2.61 |
18 |
31 |
0.00386 |
| npg-CN-0.3b |
0.39 |
0.66 |
2.14 |
0.010 |
77.8 |
2.55 |
31 |
47 |
0.00672 |
| npg-CN-0.5b |
0.36 |
0.66 |
1.78 |
0.008 |
87.1 |
2.53 |
40 |
70 |
0.01310 |
| npg-CN-0.7b |
0.35 |
0.66 |
1.88 |
0.007 |
66.7 |
2.53 |
26 |
56 |
0.00879 |
| npg-CN-1.0b |
0.34 |
0.66 |
2.21 |
0.010 |
64.8 |
2.51 |
20 |
45 |
0.00625 |
| npg-CN-0.5a |
0.38 |
0.66 |
1.93 |
0.010 |
49.2 |
2.56 |
19 |
45 |
0.00638 |
| npg-CN-0.5c |
0.33 |
0.66 |
1.71 |
0.005 |
70.8 |
2.47 |
40 |
80 |
0.01710 |
| npg-CN-0.5d |
0.12 |
0.67 |
2.13 |
0.003 |
135.1 |
2.57 |
60 |
62 |
0.01042 |
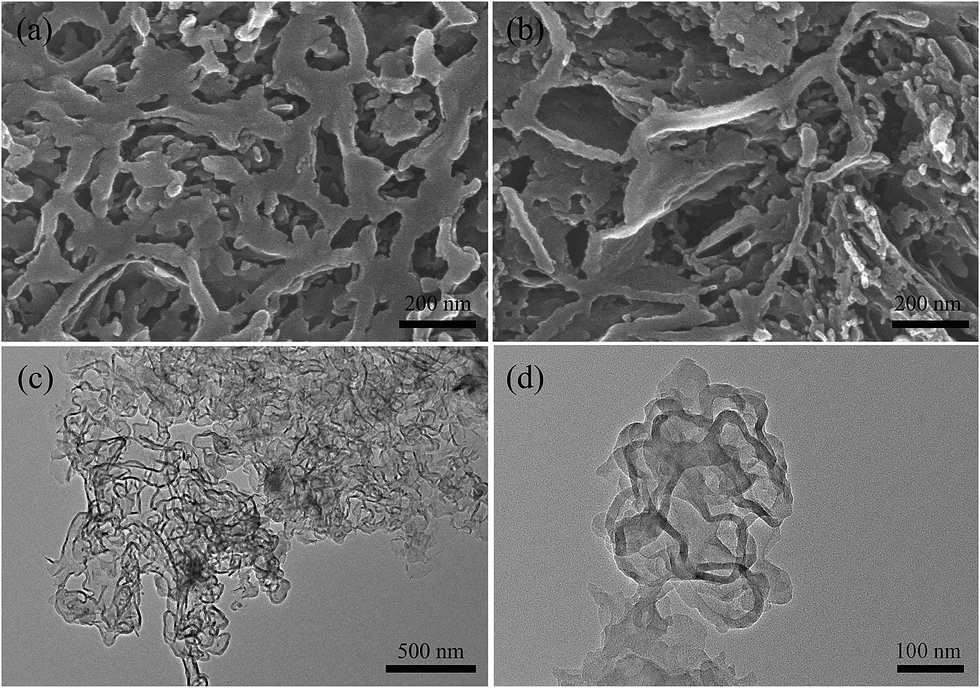
The morphology and textural structure of npg-CN were investigated by using SEM and TEM. Fig. 2a and b show typical SEM images of npg-CN-0.5c viewed from different areas. The network layers with irregular pores can be observed in Fig. 2a, and the stacking motif of the network layers can be observed in Fig. 2b. TEM images of npg-CN-0.5c with different magnifications are shown in Fig. 2c and d. Curved layers of g-CN with rolled, buckled edges interlaced with each other and formed a laver-like structure. It can conclude that irregular pore system is formed by the stacking of the porous network layers. The porosity of the npg-CN samples was further quantified by nitrogen sorption measurements. The isotherms (Fig. 3a) of all npg-CN samples exhibit a hysteresis loop range at p/p0 = 0.8–1.0, indicating a macroporous system presents in the material.27 The BET surface areas of npg-CN samples increases with increasing r value at 550 °C and to a maximum value of 87.1 m2 g−1 at r = 0.5, as shown in Table 1. Besides, with the increase of the heating temperature, the BET areas and pore volumes increases (Table S1†). Fig. 3b show the corresponding BJH pore size distribution curves of npg-CN samples determined from the adsorption branch of the isotherms. The samples of npg-CN-0.3b, 0.5b and 1.0b calcined at 550 °C show broad pore size distributions centered around 45 nm, while the samples npg-CN-0.5c and npg-CN-0.5d show broad pore size distributions centered around 60 nm. This result indicates that the pore structure will be enlarged when the heating temperature over 550 °C because the samples begin to suffer from a decomposition process,25 which can be demonstrated by the TG thermograms of MST-0.5 (Fig. S3†) and the relative low yield of npg-CN-0.5d (Table 1).
 |
| | Fig. 2 SEM images (a and b) viewed from different areas and TEM images (c and d) with different magnifications of npg-CN-0.5c. | |
 |
| | Fig. 3 (a) N2 adsorption–desorption isotherms and (b) corresponding pore size distributions of npg-CN-(0.3b, 0.5b, 1.0b, 0.5c and 0.5d). | |
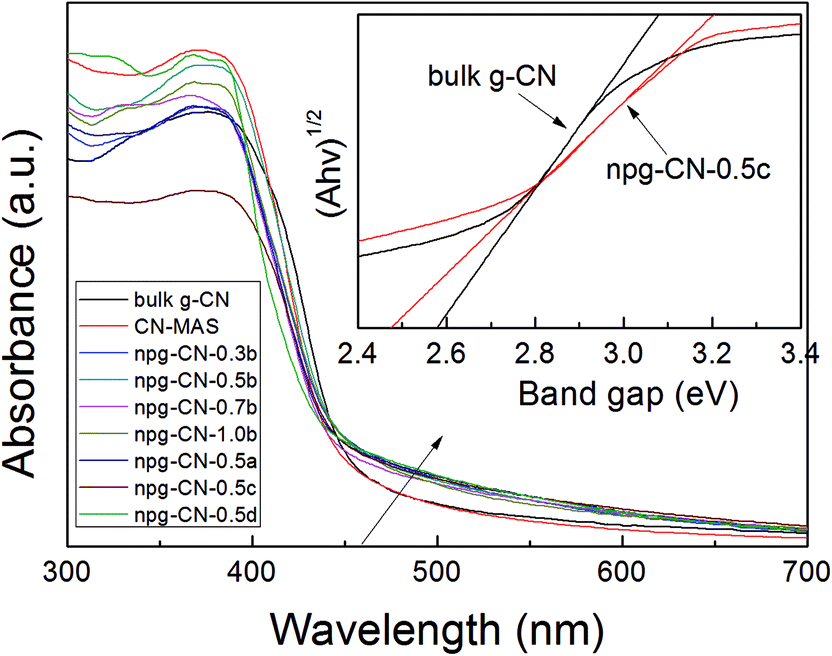
The optical absorption and energy bandgap of the npg-CN sample was examined by using UV-vis DRS measurements. In Fig. 4, the UV-vis DRS spectra of npg-CN samples were compared with that of bulk g-CN and CN–MAS. The enhanced absorption at 450–650 nm of npg-CN samples over bulk g-CN and CN–MAS results in apparent upward trailing. The corresponding band gaps (estimated from the absorption edge using Tauc plot) of bulk g-CN and npg-CN samples were summarized in Table 1, from which narrower band gaps of npg-CN samples are found than that of bulk g-CN. The npg-CN-0.5c shows the narrowest band gap, which is 0.11 eV narrower than that of bulk g-CN, as shown in Fig. 4 (inset). According to previous study,11,12 these results can be attributed to the multiple reflections of incident light within the porous structures, which increases the effective path length for light absorption, or an additional absorption of increasing number of defects derived from the nanoporous structure.28 The photoluminescence (PL) spectra excited by 340 nm of npg-CN-0.5c were compared to the spectra of bulk g-CN (Fig. S4†). The much lower fluorescence intensity of npg-CN-0.5c than that of bulk g-CN indicates that the higher separation efficiency of photogenerated electrons and holes of npg-CN samples.11
 |
| | Fig. 4 UV-vis DRS spectra and corresponding Tauc plot spectra (inset) of bulk g-CN, CN–MAS, and npg-CN samples. | |
3.2 Evaluation of photocatalytic activity of npg-CN samples
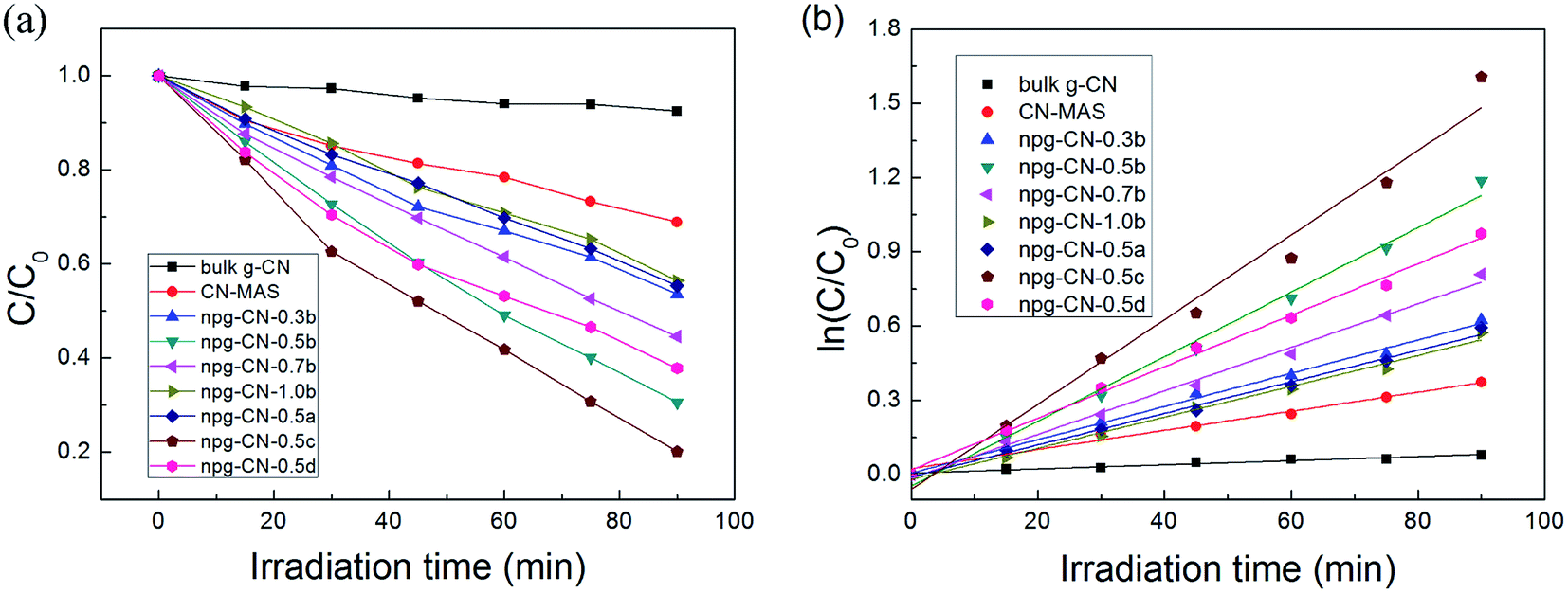
The photocatalytic activities of all the samples were evaluated for Rhodamine B (RhB) photodegradation under visible light (λ > 400 nm) irradiation. The physical adsorption properties of all samples were evaluated by stirring in the dark for 0.5 h, as shown in Fig. S5.† All samples have reached adsorption equilibrium during 10 min, and the physical adsorption ratio increased with the increase of BET area. The photodegradation curves of all the samples are shown in Fig. 5a. The bulk g-CN and the CN–MAS show poor photocatalytic degradation ratios as only 9.0% and 31% during 90 min, respectively, while all the samples of npg-CN show better activity (45–80%), as listed in Table 1. Besides, the photocatalytic activities of npg-CN samples are mostly in line with the BET areas. These results can be attributed to that the increased surface area of npg-CN can provide more catalytic active sites for photocatalytic reaction.29 Meanwhile, the kinetics of photodegradation on the catalyst surface can be well described by the first-order equation: ln(C0/C) = kt,9 where k is the reaction rate constant (min−1), as shown in Fig. 5b. The obtained reaction rate constants of all samples were listed in Table 1. The sample npg-CN-0.5c, with the narrowest band gap (2.47 eV) and relatively large BET area, exhibits the best degradation rate (0.017 min−1), which is 20 times of the bulk g-CN and 4.4 times of CN–MAS. While the sample npg-CN-0.5d, with the largest BET area, exhibits a relatively low degradation rate (0.010 min−1) because of its broad band gap (2.57 eV). These results indicate that the band gap plays an important role to the photocatalytic activity of npg-CN as well as the BET area, because narrower band gap means more effective light harvesting of the visible light under same irradiation intensity.
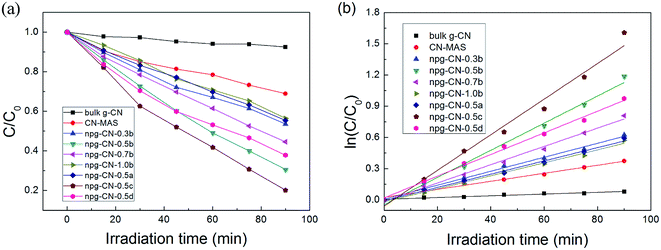 |
| | Fig. 5 (a) Photocatalytic degradation of RhB and (b) the corresponding reaction rate curves for all the samples. | |
Generally, decolorization does not mean that the dyes have been completely oxidized into harmless final products such as H2O and CO2.9 In order to determine the photodegradation of RhB is a decolorization or mineralization process, the variation of UV-vis absorption spectra of RhB degradation using npg-CN-0.5c is shown in Fig. 6. The characteristic absorption band of RhB at 554 nm decreased significantly with increasing irradiation time, accompanied by a remarkable blue-shift of the maximum absorption peak. The stepwise blue-shift of the main peak from 554 to 496 nm can be attributed to the step-by-step de-ethylation of RhB molecule,14 while the decrease in absorbance indicates the destruction of the conjugated structure. These absorption peaks completely disappeared after illumination for 5 hours, suggesting the chromophoric structure of the RhB dye can be decomposed.
 |
| | Fig. 6 UV-vis spectral changes of RhB aqueous solution in the presence of npg-CN-0.5c under visible light irradiation. | |
The stability of photocatalytic activity was investigated by using npg-CN-0.5c as the photocatalyst example (as shown in Fig. 7a). It can be seen that the photocatalytic activity of the catalyst does not show any decrease after four cycle evaluations. In addition, the XRD patterns (as shown in Fig. 7b) of the used photocatalyst after 4th cycling experiment are almost the same as that of the fresh photocatalyst, and there is no other phases in the XRD patterns. These results demonstrate that the presented npg-CN photocatalysts are stable under present reaction conditions.
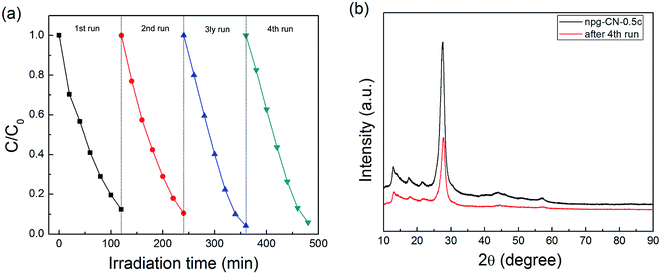 |
| | Fig. 7 (a) Cycling runs for the photocatalytic degradation of RhB in the presence of npg-CN-0.5c and (b) XRD patterns of npg-CN-0.5c before and after 4th run of photodegradation of RhB. | |
3.3 Discussion about the formation mechanism of npg-CN
The FT-IR spectra of MA, Triton X-100 and the mixture of MA and Triton X-100 (obtained by drying the mixed solution of MA and Triton X-100 (r = 0.5) directly) were compared with that of MAS and MST to demonstrate the template of Triton X-100 was totally removed. From Fig. 8a, the characteristic bands of Triton X-100 at 2800–3000 cm−1 and 1111 cm−1 can be easily found in the spectra of the mixture of MA and Triton X-100, while it cannot be found in the spectra of MST-0.5 and MAS. Meanwhile, MST-0.5 show same peaks with that of MAS, and the broad 3364 cm−1 band and 1090 cm−1 band can be attributed to the combined water and sulfate radical, respectively.30 These results indicate the Triton X-100 had been fully pre-removed in our method. The XRD patterns of MSTs were compared with that of MAS, as shown in Fig. 8b. The same peak positions indicate that MSTs have a same crystal form as MAS, corresponding to the FT-IR result. But the measured XRD peak intensity distributions for MSTs are different from the intrinsic one for MAS, indicating there are obvious crystal preferred orientations in MST samples that inferred that the polycrystalline samples of MST have nonrandom distribution of orientations31 because of the template induced effect from Triton X-100. Besides, the different concentrations of Triton X-100 can also result in different crystal preferred orientations of MST, because the shape of Triton X-100 micelles changes with the concentrations.32 As a conclusion, the mechanism of npg-CN formation was supposed as that Triton X-100 modified the process of the formation of MAS and result in a different crystal preferred orientation of melamine sulfate, which determined the final porous structures of npg-CN.
 |
| | Fig. 8 (a) FT-IR spectra of MA, Triton X-100, MAS, MST-0.5, and the mixture of MA and Triton X-100 (MA+TX-100); and (b) XRD patterns of MA, MST-0.3, MST-0.5, and MST-1.0. | |
4. Conclusions
In conclusion, the npg-CN with large surface area and low C/N ratios (0.66) was successfully prepared by directly heating Triton X-100-modified-melamine sulfate. The obtained npg-CN samples possessed high surface areas, which could be mediated by the heating temperature and the adding amount of Triton X-100. Compared with the bulk g-CN, the npg-CN samples exhibited stronger absorbance of visible light, narrower band gaps and reduced fluorescence intensity. As a result, the photocatalytic activity of npg-CN samples was greatly improved in the photodegradation of RhB with 20 times of the bulk g-CN and 4.4 times of CN–MAS at the optimum condition; meanwhile, the stability of photocatalytic activity of npg-CN samples shows better recycling life even after four cycles.
Acknowledgements
This work was financially supported by the Natural Science Foundation of China under Grant no. 10972025.
References
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- Y. Zhang, T. Mori and J. Ye, Sci. Adv. Mater., 2012, 4, 282–291 CrossRef CAS PubMed.
- A. Schwarzer, T. Saplinova and E. Kroke, Coord. Chem. Rev., 2013, 257, 2032–2062 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- Y. Li, X. Xu, P. Zhang, Y. Gong, H. Li and Y. Wang, RSC Adv., 2013, 3, 10973–10982 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. Liu, J. Huang, D. Dontosova and M. Antonietti, RSC Adv., 2013, 3, 22988–22993 RSC.
- Y. Hou, Y. Zhu, Y. Xu and X. Wang, Appl. Catal., B, 2014, 156–157, 122–127 CrossRef CAS PubMed.
- Y. Cui, Z. Ding, P. Liu, M. Antonietti, X. Fu and X. Wang, Phys. Chem. Chem. Phys., 2012, 14, 1455–1462 RSC.
- G. Dong, Z. Ai and L. Zhang, RSC Adv., 2014, 4, 5553–5560 RSC.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- J. Zhang, F. Guo and X. Wang, Adv. Funct. Mater., 2013, 23, 3008–3014 CrossRef CAS.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H.-M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed.
- S. Hu, L. Ma, J. You, F. Li, Z. Fan, F. Wang, D. Liu and J. Gui, RSC Adv., 2014, 4, 21657–21663 RSC.
- X. H. Li, X. Wang and M. Antonietti, Chem. Sci., 2012, 3, 2170–2174 RSC.
- J. Fu, B. Chang, Y. Tian, F. Xi and X. Dong, J. Mater. Chem. A, 2013, 1, 3083–3090 CAS.
- L. Huang, Y. Li, H. Xu, Y. Xu, J. Xia, K. Wang, H. Li and X. Cheng, RSC Adv., 2013, 3, 22269–22279 RSC.
- K. Dai, L. Lu, C. Liang, Q. Liu and G. Zhu, Appl. Catal., B, 2014, 156–157, 331–340 CrossRef CAS PubMed.
- S. S. Park, S. W. Chu, C. Xue, D. Zhao and C.-S. Ha, J. Mater. Chem., 2011, 21, 10801–10807 RSC.
- Y. Wang, X. Wang, M. Antonietti and Y. Zhang, ChemSusChem, 2010, 3, 435–439 CrossRef CAS PubMed.
- H. Yan, Chem. Commun., 2012, 48, 3430–3432 RSC.
- H. Yan, Y. Chen and S. Xu, Int. J. Hydrogen Energy, 2012, 37, 125–133 CrossRef CAS PubMed.
- D. M. Teter and R. J. Hemley, Science, 1996, 271, 53–55 CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- J. Xu, Y. Wang and Y. Zhu, Langmuir, 2013, 29, 10566–10572 CrossRef CAS PubMed.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 1139 CrossRef.
- Y. J. Cui, J. H. Huang, X. Z. Fu and X. C. Wang, Catal. Sci. Technol., 2012, 2, 1396–1402 CAS.
- X. H. Li, S. Z. Yang, W. D. Xiang and Q. Shi, Struct. Chem., 2007, 18, 661–666 CrossRef CAS.
- K. M. Harris, in Advanced X-Ray Crystallography, ed. K. Rissanen, Springer, Berlin, Heidelberg, 2012, vol. 315, ch. 251, pp. 133–177 Search PubMed.
- H. Song, D. Wang, X. Ma, Y. Cao and S. Zhang, Solid State Commun., 2006, 139, 430–433 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra12033g |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.