
Investigation of binap-based hydroxyphosphine arene–ruthenium(II) complexes as catalysts for nitrile hydration†
Eder Tomás-Mendivila,
Lucía Menéndez-Rodrígueza,
Javier Francosb,
Pascale Crochet a and
Victorio Cadierno*a
a and
Victorio Cadierno*a
aLaboratorio de Compuestos Organometálicos y Catálisis (Unidad Asociada al CSIC), Centro de Innovación en Química Avanzada (ORFEO-CINQA), Departamento de Química Orgánica e Inorgánica, Instituto Universitario de Química Organometálica “Enrique Moles”, Facultad de Química, Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo, Spain. E-mail: vcm@uniovi.es; Fax: +34 985103446; Tel: +34 985103453
bWestCHEM, University of Strathclyde, Department of Pure and Applied Chemistry, 295 Cathedral Street, Glasgow, G1, 1XL, UK
First published on 18th November 2014
Abstract
The binap-based hydroxyphosphine-(η6-arene)–ruthenium(II) complexes [RuX{η6:κ1(P)-PPh2-binaphthyl}{PPh2(OH)}][OTf] (X = OTf (4), Cl (5)) have been evaluated as potential catalysts for the selective hydration of nitriles to primary amides. The triflate derivative 4 proved to be the most active, being able to hydrate a large variety of aromatic, heteroaromatic, α,β-unsaturated and aliphatic nitriles in pure water at 100 °C. The utility of complex 4 to promote the catalytic rearrangement of aldoximes has also been demonstrated. In addition, insights about the role played by the hydroxyphosphine ligand PPh2(OH) during the catalytic reactions are given.
Introduction
The amide unit is present in a large number of bulk and fine chemicals, natural products, drugs, polymers, detergents and lubricants, making this functional group one of the most important in chemistry.1 Amides are typically prepared from the union of carboxylic acids and derivatives (halides, anhydrides or esters) with amines.2 However, these classical approaches are low in atom efficiency and generate large amounts of waste products. As a result of this, in 2005, the Pharmaceutical Roundtable, a forum created by the ACS Green Chemistry Institute and members of major pharmaceutical corporations worldwide, identified “amide formation avoiding poor atom economy reagents” as one of the top challenges for organic chemistry.3 In this regard, nitrile hydration represents ideally the simplest method for the sustainable preparation of primary amides (eqn (1)), but strong acids and bases combined with harsh reaction conditions have been traditionally employed to promote the process, lowering its selectivity and applicability.1,2
RC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N + H2O → RC( N + H2O → RC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)NH2 O)NH2
| (1) |
Consequently, great efforts have been devoted in both academic and industrial laboratories to the search for effective and selective nitrile hydration catalysts. In this regard, a variety of enzymes, metal oxides, metal nanoparticles and homogenous metal complexes able to promote the process under relatively mild conditions have seen the light in the most recent years, offering appealing and cleaner alternatives to the classical acid/base-based protocols.4,5 Concerning the homogeneous systems, best results have been described so far employing metal complexes in which the auxiliary ligands are directly involved in the C–O bond-forming step (cooperative/bifunctional catalysis).6 In this context, hydroxyphosphines, i.e. PR2(OH),7 have shown to be particularly useful ligands for the development of highly active nitrile hydration catalysts. First evidence was given by Ghaffar and Parkins in 1995 with the platinum(II) complex [PtH{(PMe2O)2H}{PMe2(OH)}] (1 in Fig. 1).8 Turnover frequencies (TOF) exceeding 380 h−1 and turnover numbers of up to 5700 were reported with this catalyst at 70–100 °C.9 In addition, the exquisite functional group tolerance shown by complex 1 has allowed its implementation in the synthesis of a huge number of biologically active molecules and natural products.10,11
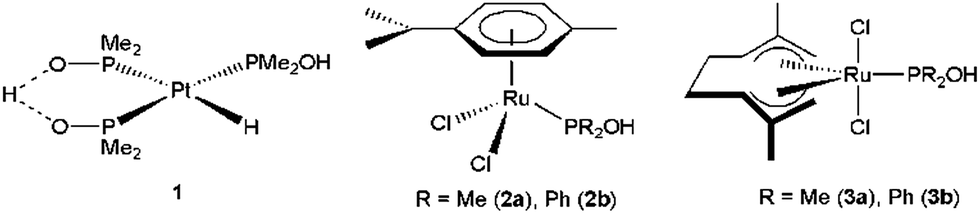 | ||
| Fig. 1 Structure of the nitrile hydration catalysts 1–3. | ||
More recently, the ruthenium complexes [RuCl2(η6-p-cymene){PMe2(OH)}] (2a in Fig. 1)12 and [RuCl2(η3:η3-C10H16){PR2(OH)}] (3a–b in Fig. 1),13 which are able to hydrate CN bonds at temperatures below 70 °C, have been described.14 Remarkably, these species proved to be active in the challenging hydration of cyanohydrins (α-hydroxynitriles), substrates very difficult to hydrate because they establish in aqueous solution an equilibrium with the corresponding carbonyl compounds and HCN. The vast majority of metal-based nitrile hydration catalysts reported to date are inactive towards cyanohydrins due to their poisoning by the cyanide anion present in the medium.15
Complexes 1–3 are also of interest from a mechanistic point of view, since two different modes of action of the hydroxyphosphine ligands R2POH have been proposed. Thus, for the platinum(II) complex 1, Ghaffar and Parkins suggested an intramolecular attack of the OH group of the phosphine on the coordinated nitrile.8,9 In this way, the metallacyclic intermediate A is initially formed, and the final amide product is subsequently generated by addition of water on the metallacycle (Scheme 1).
 | ||
| Scheme 1 Proposed mechanism for the hydration of nitriles by complex 1. | ||
In contrast, for the ruthenium complexes 2 and 3 an outer sphere mechanism, in which the direct nucleophilic attack of the water molecule on the coordinated nitrile is assisted by the hydroxyphosphine ligand via hydrogen-bonding, was proposed (transition state B in Scheme 2).12,13 This reaction pathway was supported by DFT calculations with complex [RuCl2(η6-p-cymene){PMe2(OH)}] (2a).12
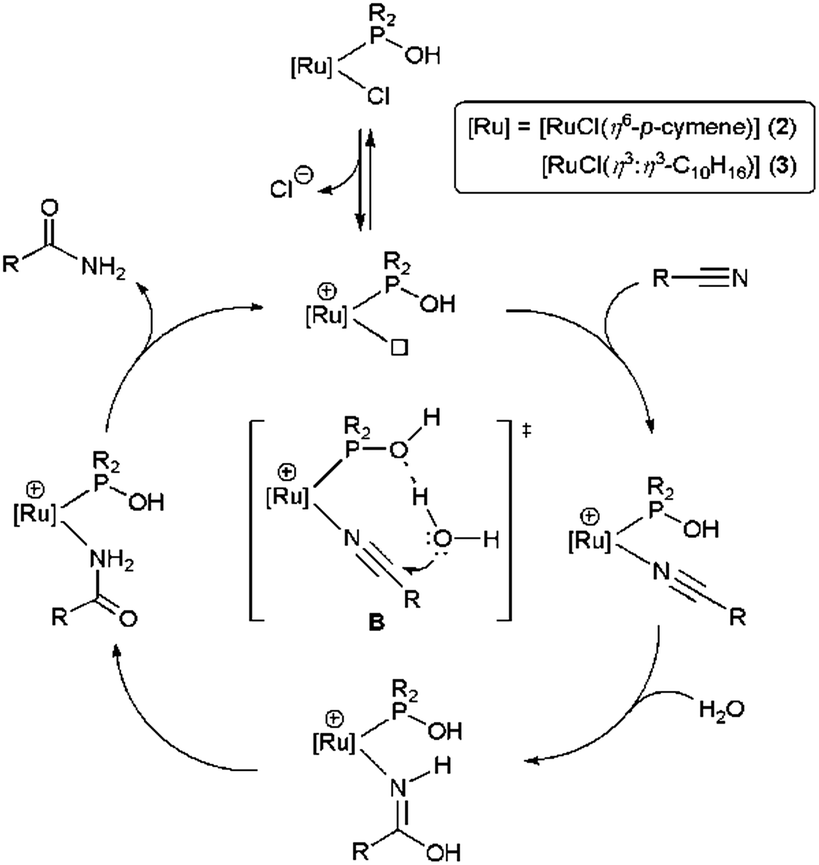 | ||
| Scheme 2 Proposed mechanism for the hydration of nitriles by complexes 2–3. | ||
During the last years our group has been heavily involved in this chemistry, developing a number of nitrile hydration catalysts based on ruthenium, osmium and rhodium complexes.6c–e,j,k,13,16 Continuing with these studies, we report herein on the catalytic behavior of the binap-based hydroxyphosphine (η6-arene)–ruthenium(II) complexes 4 and 5, whose syntheses were described some years ago by Pregosin and co-workers (Scheme 3).17 We believed that the presence a PPh2(OH) ligand, in combination with a labile triflate or chloride, may result in the discovery of new cooperative nitrile hydration catalysts. In this point, we would like to stress that, despite the relatively large number of (η6-arene)–ruthenium(II) complexes applied to date in this catalytic transformation,6c,e,h–j,12,14,16a–c,e,18 no examples of tethered derivatives have been yet reported.
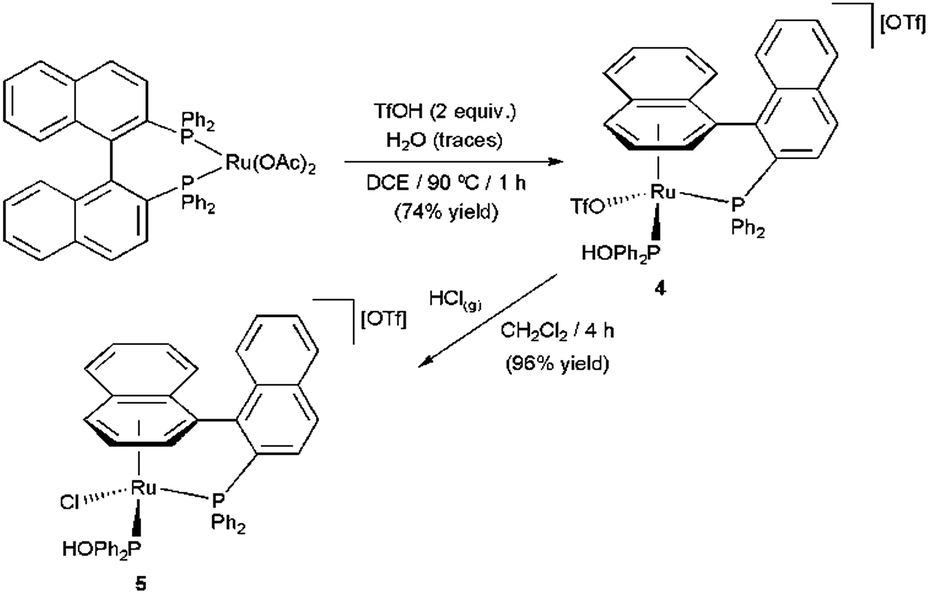 | ||
| Scheme 3 Synthetic route of the tethered (η6-arene)–ruthenium(II) complexes 4 and 5. (DCE = 1,2-dichloroethane). | ||
On the other hand, our interest in complexes 4 and 5 stemmed also from the isolation by Pregosin's group of the metallacyclic compound 6 (Fig. 2), closely related to the platinum intermediate A proposed by Ghaffar and Parkins (Scheme 1), upon treatment of 4 with an excess of 4-methylbenzonitrile.19 Consequently, the possible involvement of this type of species in the catalytic reactions has also been addressed in our study.
 | ||
| Fig. 2 Structure of the metallacyclic complex 6. | ||
Results and discussion
The catalytic potential of the tethered hydroxyphosphine–ruthenium(II) complexes 4 and 5 was initially evaluated employing benzonitrile as model substrate. As shown in Table 1, performing the hydration reactions in pure water at 100 °C and with a metal loading of 5 mol%, both complexes proved to be active (entries 1–2), providing benzamide as the unique reaction product. The triflate derivative 4 was the most effective, generating benzamide in almost quantitative yield after 5 h of heating (entry 1). The greater reactivity of 4 compared to that of 5, which required 20 h to completely consume the starting nitrile (entry 2), can be rationalized in terms of the higher lability of the triflate vs. chloride ligand,20 thus enabling a more effective generation of a vacant site on the metal for the coordination of the benzonitrile molecule. We must note in this point that the catalytic activity of complexes 4–5 is remarkably lower than that exhibited by the related diphenylhydroxyphosphine-Ru(II) derivatives [RuCl2(η6-p-cymene){PPh2(OH)}] (2b in Fig. 1) and [RuCl2(η3:η3-C10H16){PPh2(OH)}] (3b in Fig. 1) previously described in the literature.13,14 Under identical reaction conditions, both were able to convert benzonitrile into benzamide quantitatively after only 15 min (entries 3–4 vs. 1–2). The key to the exceptional activity of compounds 2–3b seems to be related to their neutral nature, since the cationic derivative [RuCl(η6-p-cymene){PPh2(OH)}(PPh3)][PF6] (7), very similar sterically and electronically to 4–5, showed also a much lower reactivity (entry 5). An easier coordination of the nitrile to the metal is expected to occur in complexes 2–3b compared to 4–5.21
|
||||
|---|---|---|---|---|
| Entry | Catalyst | t (h) | Conversionb (%) | TOFc (h−1) |
| a Reactions were performed under Ar atmosphere starting from 0.5 mmol of the corresponding nitrile (0.33 M in water).b Determined by GC (uncorrected GC areas).c Turnover frequencies ((mol product/mol Ru)/time) were calculated at the time indicated in each case.d Reaction performed with 3 mol% of complex 4.e Reaction performed with 1 mol% of complex 4.f Reaction performed at 80 °C.g Reaction performed at 60 °C. | ||||
| 1 | 4 | 5 | >99 | 4.0 |
| 2 | 5 | 20 | >99 | 1.0 |
| 3 | 2b | 0.25 | >99 | 80.0 |
| 4 | 3b | 0.25 | >99 | 80.0 |
| 5 | 7 | 18 | >99 | 1.1 |
| 6d | 4 | 8 | >99 | 4.2 |
| 7e | 4 | 24 | >99 | 4.2 |
| 8f | 4 | 24 | >99 | 0.8 |
| 9g | 4 | 24 | 77 | 0.6 |
Further studies with the triflate complex [Ru(OTf){η6:κ1(P)-PPh2-binaphthyl}{PPh2(OH)}][OTf] (4) indicated that reduction of the metal loading to 3 and 1 mol% still produced the desired benzamide in quantitative yield, although, as expected, longer reaction times were in these cases needed (entries 6–7). Conversely, the experiments carried out at lower temperatures (80 and 60 °C; entries 8–9) revealed a significant loss of activity of the complex (TOF < 1 h−1 vs. 4 h−1 at 100 °C).
The scope of 4 towards structurally diverse nitriles was subsequently explored. Thus, as summarized in Table 2, using a catalyst loading of 5 mol% and a working temperature of 100 °C, a number of aromatic (entries 1–16), heteroaromatic (entries 17–21), aliphatic (entries 22–28) and α,β-unsaturated (entry 29) nitriles could be selectively converted into the corresponding amides, generally in high yields and short times. In no case the corresponding carboxylic acids, resulting from the overhydrolysis of the amides, were detected by GC in the crude reaction mixtures. Concerning the aromatic substrates, influence of the electronic properties of the aryl rings on the activity of 4 was not observed. However, due probably to steric factors, substitution in ortho position led in general to less efficient hydrations (entry 4 vs. 5–6 or entry 11 vs. 12–13). The presence of a methylsulfanyl substituent was tolerated, but the reaction proceeded in this case much slower suggesting the competitive coordination of this group to the metal (entry 16).22
|
||||
|---|---|---|---|---|
| Entry | Nitrile | t (h) | Conversionb (%) | TOFc (h−1) |
| a Reactions were performed under Ar atmosphere starting from 0.5 mmol of the corresponding nitrile (0.33 M in water).b Determined by GC (uncorrected GC areas), isolated yields after the work-up are given in brackets.c Turnover frequencies ((mol product/mol Ru)/time) were calculated at the time indicated in each case. | ||||
| 1 | R = Ph | 5 | >99 (86) | 4.0 |
| 2 | R = 2-C6H4F | 5 | >99 (83) | 4.0 |
| 3 | R = 4-C6H4F | 5 | >99 (81) | 4.0 |
| 4 | R = 2-C6H4Cl | 7 | >99 (85) | 2.9 |
| 5 | R = 3-C6H4Cl | 5 | >99 (83) | 4.0 |
| 6 | R = 4-C6H4Cl | 5 | >99 (79) | 4.0 |
| 7 | R = 3-C6H4Br | 5 | >99 (82) | 4.0 |
| 8 | R = 4-C6H4Br | 5 | >99 (80) | 4.0 |
| 9 | R = 3-C6H4NO2 | 5 | >99 (84) | 4.0 |
| 10 | R = C6F5 | 5 | >99 (83) | 4.0 |
| 11 | R = 2-C6H4Me | 24 | 86 (70) | 0.7 |
| 12 | R = 3-C6H4Me | 5 | >99 (84) | 4.0 |
| 13 | R = 4-C6H4Me | 5 | >99 (80) | 4.0 |
| 14 | R = 4-C6H4OMe | 5 | 99 (82) | 4.0 |
| 15 | R = 4-C6H4OH | 5 | >99 (78) | 4.0 |
| 16 | R = 4-C6H4SMe | 24 | 90 (73) | 0.8 |
| 17 | R = 2-pyridyl | 24 | 36 (10) | 0.3 |
| 18 | R = 3-pyridyl | 3 | >99 (82) | 6.7 |
| 19 | R = 4-pyridyl | 3 | >99 (81) | 6.7 |
| 20 | R = 3-thienyl | 10 | >99 (78) | 2.0 |
| 21 | R = 5-Me-2-furyl | 3 | 99 (79) | 6.7 |
| 22 | R = Me | 5 | >99 (83) | 4.0 |
| 23 | R = n-C6H13 | 6 | >99 (86) | 3.3 |
| 24 | R = Cy | 24 | 91 (74) | 0.8 |
| 25 | R = CH2Cl | 5 | >99 (80) | 4.0 |
| 26 | R = CHCl2 | 4 | >99 (86) | 5.0 |
| 27 | R = CCl3 | 4 | >99 (82) | 5.0 |
| 28 | R = CH2OPh | 4 | 98 (79) | 4.9 |
| 29 | R = (E)-CHCHPh |
6 | 99 (83) | 3.3 |

With the exception of 2-pyridinecarbonitrile (entry 17) and 3-thiophenecarbonitrile (entry 20), fast conversions of different heteroaromatic nitriles into the corresponding amides were also achieved using complex [Ru(OTf){η6:κ1(P)-PPh2-binaphthyl}{PPh2(OH)}][OTf] (4) (entries 17–21). Catalyst inhibition by strong chelate coordination of the picolinamide formed in the medium, an observation previously quoted in the literature,6f,i,18f,23 could explain the comparatively longer time required for the hydration of 2-pyridinecarbonitrile. On the other hand, the sensitivity of 4 to the steric demand of the substrates was again evidenced in the hydration of aliphatic nitriles (entries 22–28), where fast reactions were in general observed except for the bulky cyclohexanecarbonitrile (entry 24).
Worthy of note, the synthetic utility of [Ru(OTf){η6:κ1(P)-PPh2-binaphthyl}{PPh2(OH)}][OTf] (4) is not restricted to the catalytic hydration of nitriles. Thus, as shown in Table 3, this complex is also able to promote the rearrangement of aldoximes, an alternative atom economical process for forming primary amides which involves a dehydration/hydration sequence via the corresponding nitrile intermediate.24 Using the same reaction conditions, i.e. 5 mol% of 4 in pure water at 100 °C, the amide products could be generated in >89% GC-yield in times similar to those required for the hydration of the respective nitriles (see Table 2).
|
||||
|---|---|---|---|---|
| Entry | Aldoxime | t (h) | Conversionb (%) | TOFc (h−1) |
| a Reactions were performed under Ar atmosphere starting from 0.5 mmol of the corresponding aldoxime (0.33 M in water).b Determined by GC (uncorrected GC areas), isolated yields after the work-up are given in brackets.c Turnover frequencies ((mol product/mol Ru)/time) were calculated at the time indicated in each case. | ||||
| 1 | R = Ph | 6 | 98 (85) | 3.2 |
| 2 | R = 4-C6H4F | 6 | >99 (87) | 3.3 |
| 3 | R = 4-C6H4Cl | 6 | >99 (83) | 3.3 |
| 4 | R = 4-C6H4Me | 6 | >99 (81) | 3.3 |
| 5 | R = 4-C6H4OMe | 6 | 97 (79) | 3.2 |
| 6 | R = n-C6H13 | 6 | >99 (82) | 3.3 |
| 7 | R = Cy | 24 | 89 (70) | 0.7 |
| 8 | R = (E)-CHCHPh |
7 | 98 (81) | 2.8 |

As commented in the introduction section, Pregosin's group reported that treatment of complex 4 with an excess 4-methylbenzonitrile, in DCE at 70 °C for 2 days, leads to the clean and quantitative formation of the metallacyclic derivative 6 (Fig. 2). This compound results from the partial displacement of the tethered binaphthyl-phosphine (η6 to η2 coordination) and the triflate ligand by three molecules of the nitrile, and subsequent addition of the OH group of the hydroxyphosphine on one of the coordinated nitriles.19 The structure of complex 6, proposed by Pregosin and co-workers on the basis of NMR and mass spectroscopic data, has now been fully confirmed by us through a single-crystal X-ray diffraction study. Fig. 3 shows an ORTEP view of the dication, in which the ruthenium atom is in a distorted octahedral environment (selected bonding parameters are listed in the caption). As expected, the two 4-methylbenzonitrile molecules, mutually trans disposed, are bound to ruthenium in a nearly linear fashion (Ru–N–C angles of 170.0(3) and 176.8(3)°) with standard Ru–N and N–C distances.25 In the structure, the binaphthyl-phosphine adopts a η2:κ1(P) coordination mode, the relatively long Ru(1)–C(31) and Ru(1)–C(40) bond lengths observed (2.740(4) and 2.557(3) Å, respectively) suggesting a very weak ruthenium–olefin interaction.26 The C(31)–C(40) distance of 1.387(5) Å, which is only slightly longer than an uncomplexed double bond (ca. 1.34 Å), is in complete accord with this statement. Finally, the bond lengths within the –Ru–P–O–CN– five-membered ring showed the expected values, the most remarkable feature of this unit being the relatively small P(1)–Ru(1)–N(1) angle (79.38(8)°). This value reflects some strain within the metallacycle.
 | ||
| Fig. 3 ORTEP-type view of the structure of complex 6 showing the crystallographic labelling scheme. Hydrogen atoms, except that on N(1), and triflate anions have been omitted for clarity. Thermal ellipsoids are drawn at 30% probability level. Selected bond lengths (Å): Ru(1)–P(1) 2.2267(8); Ru(1)–P(2) 2.3352(8); Ru(1)–N(1) 2.120(3); Ru(1)–N(2) 2.005(3); Ru(1)–N(3) 2.024(3); Ru(1)–C(31) 2.740(4); Ru(1)–C(40) 2.557(3); Ru–C* 2.5576(2); C(31)–C(40) 1.387(5); P(1)–O(1) 1.670(2); C(1)–O(1) 1.360(4); C(1)–N(1) 1.281(4); N(2)–C(53) 1.138(4); N(3)–C(61) 1.138(4). Selected bond angles (°): P(1)–Ru(1)–P(2) 99.48(3); P(1)–Ru(1)–N(1) 79.38(8); P(1)–Ru(1)–N(2) 86.60(8); P(1)–Ru(1)–N(3) 95.64(8); P(1)–Ru(1)–C* 173.91(2); P(2)–Ru(1)–N(1) 175.53(8); P(2)–Ru(1)–N(2) 99.03(8); P(2)–Ru(1)–N(3) 87.47(8); P(2)–Ru(1)–C* 85.65(2); N(1)–Ru(1)–N(2) 85.24(10); N(1)–Ru(1)–N(3) 88.4(1); N(1)–Ru(1)–C* 95.74(8); N(2)–Ru(1)–N(3) 172.71(10); N(2)–Ru(1)–C* 89.39(7); N(3)–Ru(1)–C* 87.80(7); Ru(1)–P(1)–O(1) 103.21(8); Ru(1)–N(1)–C(1) 119.7(2); N(1)–C(1)–O(1) 118.8(3); C(1)–O(1)–P(1) 117.6(2); Ru(1)–N(2)–C(53) 170.0(3), Ru(1)–N(3)–C(61) 176.8(3). C* denotes the centroid of the olefinic C(31)–C(40) bond. | ||
To determine whether metallacyclic species related to 6 are involved as intermediates in the catalytic nitrile hydration reactions promoted by 4, we first studied the behaviour of complex 6 towards water. Experiments conducted in a NMR tube (acetone-d6 solution) showed that, in the presence of an excess of water at 70 °C, 6 readily evolves to generate cleanly and quantitatively the known aquo-complex 8 (ref. 19) and 4-methylbenzamide (Scheme 4). Formation of 8 was also observed at room temperature, but an incomplete reaction took place at this temperature even after 16 h (details are given in the ESI† file).
 | ||
| Scheme 4 The reactivity of complex 6 towards water. | ||
Based on these observations, a catalytic cycle related to that suggested by Ghaffar and Parkins for the platinum(II) complex [PtH{(PMe2O)2H}{PMe2(OH)}] (1) could be envisaged (Scheme 5). However, this reaction pathway does not appear to be the predominant one since, compared to 4, the isolated complexes 6 and 8 showed a lower activity in the hydration of 4-methylbenzonitrile. Thus, under identical reaction conditions, i.e. with a metal loading of 5 mol% in pure water at 100 °C, 7 hours were in both cases needed to achieve the complete transformation of 4-methylbenzonitrile into 4-methylbenzamide (TOF = 2.8 h−1 vs. 4.0 h−1 in the case of 4; entry 13 in Table 2). We assume, therefore, that complex [Ru(OTf){η6:κ1(P)-PPh2-binaphthyl}{PPh2(OH)}][OTf] (4) operates mainly through an outer sphere mechanism analogous to that proposed for the related species [RuCl2(η6-p-cymene){PMe2(OH)}] (2a) and [RuCl2(η3:η3-C10H16){PR2(OH)}] (3a–b) (Scheme 2).
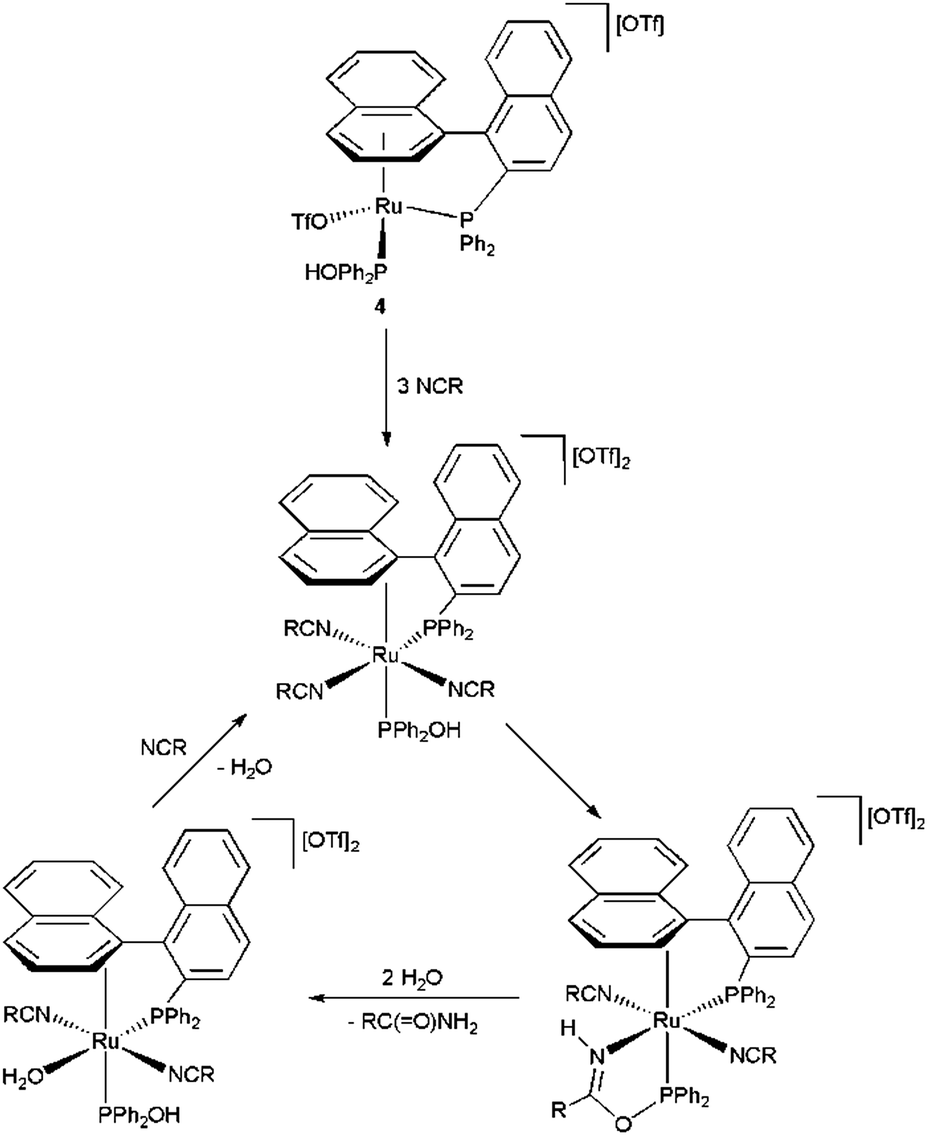 | ||
| Scheme 5 A possible catalytic cycle for the hydration of nitriles by complex 4. | ||
Conclusions
The catalytic hydration of nitriles is an important transformation in many chemical and pharmaceutical processes because it provides an atom economical route to amides. In this context, ruthenium complexes have emerged in recent years as the most versatile nitrile hydration catalysts with regard to activity, selectivity, and tolerance to functional groups.4j,k Our study contributes to the field with the discovery of new selective and broad scope catalysts, namely [RuX{η6:κ1(P)-PPh2-binaphthyl}{PPh2(OH)}][OTf] (X = OTf (4), Cl (5)). These species, whose activity is based on the presence of a cooperative hydroxyphosphine ligand, represent the first examples of tethered (η6-arene)–ruthenium(II) complexes able to promote this synthetically useful transformation. Although the activities found do not exceed that of the neutral species [RuCl2(η6-p-cymene){PPh2(OH)}] and [RuCl2(η3:η3-C10H16){PPh2(OH)}] previously described in the literature, the good conversions observed support the exploration of other ruthenium complexes with PR2(OH) ligands in this catalytic reaction.Experimental
General methods
The manipulations were performed under argon atmosphere using vacuum-line and standard Schlenk or sealed-tube techniques. All reagents were obtained from commercial suppliers and used as received, with the exception of complexes [{RuCl(μ-Cl) (η6-p-cymene)}2],27 3b,13 4,17b 5,17c 6 (ref. 19) and 8,19 Ph2P(O)H,28 and the aldoximes included in Table 3,24 which were prepared following the methods previously reported in the literature. GC measurements were performed on a Hewlett-Packard HP6890 equipment using a Supelco Beta-Dex™ 120 column (30 m length; 250 μm diameter). Infrared spectra were recorded on a Perkin-Elmer 1720-XFT spectrometer. NMR spectra were recorded on Bruker DPX-300 or AV400 instruments. The chemical shift values (δ) are given in parts per million and are referred to the residual peak of the deuterated solvent employed (1H and 13C) or to an external 85% aqueous H3PO4 solution (31P). DEPT experiments have been carried out for all the compounds reported. Elemental analyses were provided by the Analytical Service of the Instituto de Investigaciones Químicas (IIQ-CSIC) of Seville.
Synthesis of [RuCl2(η6-p-cymene){PPh2(OH)}] (2b)14
The dimeric precursor [{RuCl(μ-Cl)(η6-p-cymene)}2] (0.1 g, 0.16 mmol) was partially dissolved in 15 mL of THF and a slight excess of Ph2P(O)H (0.073 g, 0.36 mmol) was added. The reaction mixture was stirred at room temperature overnight. The solvent was then removed under reduced pressure, and the residue dissolved in the minimum amount of CH2Cl2 (ca. 3 mL). Complex 2b was isolated from the solution by adding hexanes (ca. 30 mL) and washing of the resulting orange solid precipitate with hexanes (2 × 10 mL) and diethyl ether (2 × 5 mL). Yield: 0.155 g (95%). IR (KBr): ν = 3391 (br), 3043 (m), 2962 (w), 1435 (s), 1378 (w), 1109 (vs), 1027 (w), 866 (s), 857 (m), 759 (w), 747 (m), 713 (m), 698 (s), 528 (vs), 498 (s), 469 (m) cm−1. 31P{1H} NMR (CDCl3): δ = 107.0 (s) ppm. 1H NMR (CDCl3): δ = 1.00 (d, 6H, 3JHH = 6.9 Hz, CHMe2), 2.04 (s, 3H, Me), 2.54 (sept, 1H, 3JHH = 6.9 Hz, CHMe2), 5.28 (d, 2H, 3JHH = 5.6 Hz, CH of cym), 5.42 (d, 2H, 3JHH = 5.6 Hz, CH of cym), 7.50 (m, 6H, Ph), 7.74 (m, 4H, Ph) ppm; OH signal not observed. 13C{1H} NMR (CDCl3): δ = 17.8 (s, Me), 21.7 (s, CHMe2), 30.3 (s, CHMe2), 86.8 (d, 2JPC = 5.7 Hz, CH of cym), 89.6 (d, 2JPC = 4.8 Hz, CH of cym), 96.6 (s, C of cym), 108.6 (s, C of cym), 128.2 (d, 3JPC = 10.7 Hz, m-Ph), 131.2 (s, p-Ph), 131.6 (d, 2JPC = 11.6 Hz, o-Ph), 137.2 (d, 1JPC = 58.0 Hz, i-Ph) ppm. Elemental analysis calcd (%) for C22H25Cl2OPRu: C 51.98, H 4.96; found: C 51.90, H 5.12.
Synthesis of [RuCl(η6-p-cymene){PPh2(OH)}(PPh3)][PF6] (7)
To a solution of complex [RuCl2(η6-p-cymene){PPh2(OH)}] (2b) (0.1 g, 0.197 mmol) in 10 mL of methanol were added NaPF6 (0.034 g, 0.2 mmol) and PPh3 (0.052 g, 0.2 mmol), and the resulting mixture stirred at room temperature for 5 h. The solvent was then removed under vacuum, the crude product extracted with CH2Cl2 (20 mL), and the extract filtered over Kieselguhr. Concentration of the resulting solution to ca. 3 mL followed by the addition of 30 mL of diethyl ether precipitated a yellow solid, which was washed twice with 5 mL of diethyl ether and dried in vacuo. Yield: 0.144 g (83%). IR (KBr): ν = 3369 (br), 3056 (m), 2965 (w), 1590 (m), 1482 (m), 1437 (s), 1261 (w), 1130 (m), 1096 (s), 839 (vs), 747 (m), 695 (s), 527 (s), 523 (s), 496 (m) cm−1. 31P{1H} NMR (CD2Cl2): δ = −144.3 (sept, JFP = 718.2 Hz, PF6−), 26.8 (d, 2JPP = 57.8 Hz, PPh3), 99.6 (br, PPh2OH) ppm. 1H NMR (CD2Cl2): δ = 1.12 (d, 3H, 3JHH = 7.6 Hz, CHMe2), 1.23 (s, 3H, Me), 1.28 (d, 3H, 3JHH = 7.8 Hz, CHMe2), 2.66 (m, 1H, CHMe2), 4.69 (s, 1H, CH of cym), 4.81 (d, 1H, 3JHH = 6.6 Hz, CH of cym), 5.83 (br, 1H, CH of cym), 5.97 (d, 1H, 3JHH = 6.6 Hz, CH of cym), 7.35–8.11 (m, 25H, Ph) ppm; OH signal not observed. 13C{1H} NMR (CD2Cl2): δ = 16.2 (s, Me), 21.0 (s, CHMe2), 21.7 (s, CHMe2), 31.2 (s, CHMe2), 90.5 (s, CH of cym), 90.7 (s, CH of cym), 91.9 (s, CH of cym), 92.1 (s, CH of cym), 99.4 (s, C of cym), 100.5 (s, C of cym), 128.4 (d, 3JPC = 9.8 Hz, m-Ph), 128.8 (d, 3JPC = 9.7 Hz, m-Ph), 129.3 (d, 1JPC = 46.4 Hz, i-Ph), 130.3 (d, 2JPC = 10.9 Hz, o-Ph), 130.9 (s, p-Ph), 131.0 (s, p-Ph), 132.3 (d, 1JPC = 43.3 Hz, i-Ph), 134.2 (d, 2JPC = 11.3 Hz, o-Ph) ppm. Elemental analysis calcd (%) for C40H40F6P3ClORu: C 54.58, H 4.58; found: C 54.49, H 4.61.General procedure for the catalytic hydration of nitriles with [Ru(OTf){η6:κ1(P)-PPh2-binaphthyl}{PPh2(OH)}][OTf] (4)
The corresponding nitrile (0.5 mmol), water (1.5 mL), and the ruthenium complex 4 (0.026 g, 0.025 mmol) were introduced into a Teflon-capped sealed tube, and the reaction mixture stirred at 100 °C for the indicated time (see Table 2). The course of the reaction was monitored regularly taking samples of ca. 20 μL, which, after extraction with CH2Cl2 (3 mL), were analyzed by GC. After elimination of the solvent under reduced pressure, the crude reaction mixture was purified by column chromatography over silica gel using CH2Cl2 as the eluent. The identity of the resulting amides was assessed by comparison of their NMR spectroscopic data with those reported in the literature.General procedure for the catalytic rearrangement of aldoximes with [Ru(OTf){η6:κ1(P)-PPh2-binaphthyl}{PPh2(OH)}][OTf] (4)
The corresponding aldoxime (0.5 mmol), water (1.5 mL), and the ruthenium complex 4 (0.026 g, 0.025 mmol) were introduced into a Teflon-capped sealed tube, and the reaction mixture stirred at 100 °C for the indicated time (see Table 3). The course of the reaction was monitored regularly taking samples of ca. 20 μL, which, after extraction with CH2Cl2 (3 mL), were analyzed by GC. After elimination of the solvent under reduced pressure, the crude reaction mixture was purified by column chromatography over silica gel using CH2Cl2 as the eluent. The identity of the resulting amides was assessed by comparison of their NMR spectroscopic data with those reported in the literature.X-ray crystal structure determination of compound 6
Crystals of 6 suitable for X-ray diffraction analysis were obtained by slow diffusion of Et2O into a saturated solution of the complex in CH2Cl2. The most relevant crystal and refinement data are collected in Table 4. Data collection was performed with an Oxford Diffraction Xcalibur single crystal diffractometer using Mo-Kα radiation (λ = 0.71073 Å). Images were collected at a fixed crystal-detector distance of 45 mm, using the oscillation method with 1° oscillation and 10.47 s variable exposure time per image. Data collection strategy was calculated with the program CrysAlis Pro CCD.29 Data reduction and cell refinement was performed with the program CrysAlis Pro RED.29 An empirical absorption correction was applied using the SCALE3 ABSPACK algorithm as implemented in the program CrysAlis Pro RED.29 The software package WINGX30 was used for space group determination, structure solution and refinement. The structure was solved by direct methods using SIR2004.31 Isotropic least-squares refinement on F2 using SHELXL97 was performed.32 During the final stages of the refinements, all the positional parameters and the anisotropic temperature factors of all the non-H atoms were refined. The H atoms were geometrically located and their coordinates were refined riding on their parent atoms. Atoms H1n and H40 were found from the Fourier map and included in a refinement with isotropic parameters. The maximum residual electron density is located near to heavy atoms. The function minimized was {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2 where w = 1/[σ2(Fo2) + (0.0524P)2 + 7.6561P] with σ(Fo2) from counting statistics and P = (Max (Fo2,0) + 2Fc2)/3. Atomic scattering factors were taken from the International Tables for X-ray crystallography.33 Geometrical calculations were made with PARST.34 The crystallographic plots were made with ORTEP-3.35| a R1 = ∑(|Fo| − |Fc|)/∑|Fo|; wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. | |
|---|---|
| Empirical formula | C70H55O7F6N3P2S2Ru |
| Formula weight | 1391.30 |
| Temperature/K | 124 |
| Wavelength/Å | 0.71073 |
| Crystal system | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Crystal size/mm | 0.25 × 0.10 × 0.05 |
| a/Å | 11.4467(3) |
| b/Å | 15.33007(6) |
| c/Å | 17.6643(6) |
| α (°) | 83.794(3) |
| β (°) | 87.765(2) |
| γ (°) | 86.541(3) |
| Z | 2 |
| Volume/Å3 | 3074.4(2) |
| Calculated density/g cm−3 | 1.503 |
| μ/mm−1 | 0.453 |
| F(000) | 1424 |
| θ range/° | 2.97–28.46 |
| Index ranges | −15 ≤ h ≤ 15 |
| −20 ≤ k ≤ 20 | |
| −21 ≤ l ≤ 23 | |
| Completeness to θmax | 91.5% |
| No. of reflns. Collected | 14221 |
| No. of unique reflns. | 11303 (Rint = 0.100) |
| No. of parameters/restraints | 828/0 |
| Refinement method | Full-matrix least-squares on F2 |
| Goodness-of-fit on F2 | 1.035 |
| R1 [I > 2σ(I)]a | 0.0519 |
| wR2 [I > 2σ(I)]a | 0.0717 |
| R1 (all data) | 0.1204 |
| R2 (all data) | 0.1323 |
| Largest diff. Peak and hole/e Å−3 | 1.705 and −1.764 |
Acknowledgements
This work was financially supported by MINECO (Projects CTQ2010-14796 and CTQ2013-40591) of Spain. E.T.-M. and L.M.-R. thank MECD and MINECO of Spain and the European Social Fund (ESF) for the award of FPU and FPI fellowships, respectively.Notes and references
- See, for example: (a) The Chemistry of Amides, ed. J. Zabicky, Wiley-Interscience, New York, 1970 Search PubMed; (b) The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science, ed. A. Greenberg, C. M. Breneman and J. F. Liebman, John Wiley & Sons, New York, 2000 Search PubMed; (c) Polyesters and Polyamides, ed. B. L. Deopura, B. Gupta, M. Joshi and R. Alagirusami, CRC Press, Boca Raton, 2008 Search PubMed; (d) I. Johansson, in Kirk-Othmer Encyclopedia of Chemical Technology, JohnWiley & Sons, New York, 2004, vol. 2, pp. 442–463 Search PubMed.
- See, for example: (a) Methoden Org. Chem. (Houben Weyl), ed. D. Dopp and H. Dopp, Thieme Verlag, Stuttgart, 1985, vol. E5(2), pp. 1024–1031 Search PubMed; (b) P. D. Bailey, T. J. Mills, R. Pettecrew and R. A. Price, in Comprehensive Organic Functional Group Transformations II, ed. A. R. Katritzky and R. J. K. Taylor, Elsevier, Oxford, 2005, vol. 5, pp. 201–294 Search PubMed; (c) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS PubMed; (d) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
- (a) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC; (b) For a related article highlighting this point, see: J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC.
- For selected review articles, see: (a) M. Kobayashi and S. Shimizu, Curr. Opin. Chem. Biol., 2000, 4, 95 CrossRef CAS; (b) V. Y. Kukushkin and A. J. L. Pombeiro, Chem. Rev., 2002, 102, 1771 CrossRef CAS PubMed; (c) P. K. Mascharak, Coord. Chem. Rev., 2002, 225, 201 CrossRef CAS; (d) J. A. Kovacs, Chem. Rev., 2004, 104, 825 CrossRef CAS PubMed; (e) V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1 CrossRef CAS PubMed; (f) S. Prasad and T. C. Bhalla, Biotechnol. Adv., 2010, 28, 725 CrossRef CAS PubMed; (g) T. J. Ahmed, S. M. M. Knapp and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 949 CrossRef CAS PubMed; (h) R. García-Álvarez, P. Crochet and V. Cadierno, Green Chem., 2013, 15, 46 RSC; (i) E. L. Downs and D. R. Tyler, Coord. Chem. Rev., 2014, 280, 28 CrossRef CAS PubMed; (j) P. Crochet and V. Cadierno, Top. Organomet. Chem., 2014, 48, 81 CrossRef; (k) R. García-Álvarez, J. Francos, E. Tomás-Mendivil, P. Crochet and V. Cadierno, J. Organomet. Chem., 2014, 771, 93 CrossRef PubMed.
- We must note that enzymes have already found application in the commercial production of some relevant amides, such as acrylamide, nicotinamide, and the antiepileptic drug levetiracetam. See, for example: (a) H. Yamada and M. Kobayashi, Biosci., Biotechnol., Biochem., 1996, 60, 1391 CrossRef CAS PubMed; (b) J. Tao and J.-H. Xu, Curr. Opin. Chem. Biol., 2009, 13, 43 CrossRef CAS PubMed; (c) S. Sanchez and A. L. Demain, Org. Process Res. Dev., 2011, 15, 224 CrossRef CAS; (d) B. Li, J. Su and J. Tao, Org. Process Res. Dev., 2011, 15, 291 CrossRef CAS.
- For selected examples, see: (a) W. K. Fung, X. Huang, M. L. Man, S. M. Ng, M. Y. Hung, Z. Lin and C. P. Lau, J. Am. Chem. Soc., 2003, 125, 11539 CrossRef CAS PubMed; (b) T. Oshiki, H. Yamashita, K. Sawada, M. Utsunomiya, K. Takahashi and K. Takai, Organometallics, 2005, 24, 6287 CrossRef CAS; (c) V. Cadierno, J. Francos and J. Gimeno, Chem.–Eur. J., 2008, 14, 6601 CrossRef CAS PubMed; (d) V. Cadierno, J. Díez, J. Francos and J. Gimeno, Chem.–Eur. J., 2010, 16, 9808 CrossRef CAS PubMed; (e) R. García-Álvarez, J. Díez, P. Crochet and V. Cadierno, Organometallics, 2011, 30, 5442 CrossRef; (f) W.-C. Lee and B. J. Frost, Green Chem., 2012, 14, 62 RSC; (g) P. Daw, A. Sinha, S. M. W. Rahaman, S. Dinda and J. K. Bera, Organometallics, 2012, 31, 3790 CrossRef CAS; (h) S. M. M. Knapp, T. J. Sherbow, R. B. Yelle, L. N. Zakharov, J. J. Juliette and D. R. Tyler, Organometallics, 2013, 32, 824 CrossRef CAS; (i) W.-C. Lee, J. M. Sears, R. A. Enow, K. Eads, D. A. Krogstad and B. J. Frost, Inorg. Chem., 2013, 52, 1737 CrossRef CAS PubMed; (j) R. García-Álvarez, M. Zablocka, P. Crochet, C. Duhayon, J.-P. Majoral and V. Cadierno, Green Chem., 2013, 15, 2447 RSC; (k) E. Tomás-Mendivil, R. García-Álvarez, C. Vidal, P. Crochet and V. Cadierno, ACS Catal., 2014, 4, 1901 CrossRef.
- The chemistry and catalytic applications of these ligands, also refereed to as phosphinites or phosphinous acids, have been reviewed: (a) L. Ackermann, Synthesis, 2006, 1557 CrossRef CAS; (b) T. M. Shaikh, C.-M. Weng and F.-E. Hong, Coord. Chem. Rev., 2012, 256, 771 CrossRef CAS PubMed.
- T. Ghaffar and A. W. Parkins, Tetrahedron Lett., 1995, 36, 8657 CrossRef CAS.
- T. Ghaffar and A. W. Parkins, J. Mol. Catal. A: Chem., 2000, 160, 249 CrossRef CAS.
- See, for example: (a) J. Akisanya, A. W. Parkins and J. W. Steed, Org. Process Res. Dev., 1998, 2, 274 CrossRef CAS; (b) X.-B. Jiang, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Org. Chem., 2004, 69, 2327 CrossRef CAS PubMed; (c) T. J. Greshock and R. L. Funk, Org. Lett., 2006, 8, 2643 CrossRef CAS PubMed; (d) X. Jiang, N. Williams and J. K. De Brabander, Org. Lett., 2007, 9, 227 CrossRef CAS PubMed; (e) B. Wang, F. Wu, Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2007, 129, 768 CrossRef CAS PubMed; (f) T. Kan, Y. Kawamoto, T. Asakawa, T. Furuta and T. Fukuyama, Org. Lett., 2008, 10, 169 CrossRef CAS PubMed; (g) R. A. Jones and M. J. Krische, Org. Lett., 2009, 11, 1849 CrossRef CAS PubMed; (h) L. E. Brown, Y. R. Landaverry, J. R. Davies, K. A. Milinkevich, S. Ast, J. S. Carlson, A. G. Oliver and J. P. Konopelski, J. Org. Chem., 2009, 74, 5405 CrossRef CAS PubMed; (i) F. D. J. Cortez and R. Sarpong, Org. Lett., 2010, 12, 1428 CrossRef PubMed; (j) C.-K. Mai, M. F. Sammons and T. Sammakia, Angew. Chem., Int. Ed., 2010, 49, 2397 CrossRef PubMed; (k) T. A. Brugel, R. W. Smith, M. Balestra, C. Becker, T. Daniels, G. M. Koether, S. R. Throner, L. M. Panko, D. G. Brown, R. Liu, J. Gordon and M. F. Peters, Bioorg. Med. Chem. Lett., 2010, 20, 5405 CrossRef CAS PubMed; (l) M. K. M. Tun, D.-J. Wüstmann and S. B. Herzon, Chem. Sci., 2011, 2, 2251 RSC; (m) L. Yao, B. Pitta, P. C. Ravikumar, M. Purzycki and F. F. Fleming, J. Org. Chem., 2012, 77, 3651 CrossRef CAS PubMed; (n) R. S. Andrews, J. L. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2012, 51, 4140 CrossRef PubMed.
- For a recent study on related platinum complexes, see: S. M. M. Knapp, T. J. Sherbow, T. J. Ahmed, I. Thiel, L. N. Zakharov, J. J. Juliette and D. R. Tyler, J. Inorg. Organomet. Polym., 2014, 24, 145 CrossRef CAS.
- S. M. M. Knapp, T. J. Sherbow, R. B. Yelle, J. J. Juliette and D. R. Tyler, Organometallics, 2013, 32, 3744 CrossRef CAS.
- E. Tomás-Mendivil, F. J. Suárez, J. Díez and V. Cadierno, Chem. Commun., 2014, 50, 9661 RSC.
- The utility of related [RuX2(η6-arene){PR2(OH)}] (arene = C6H6, p-cymene, C6Me6; X = Cl, Br, I; R = Ph, nBu; not all combinations) complexes, including 2b (Fig. 1) whose synthesis and characterization are fully described here, for nitrile hydration reactions has been the subject of a recent patent: T. Oshiki and M. Muranaka, PCT Int. Appl., WO 2012/017966, 2012.
- (a) T. J. Ahmed, B. R. Fox, S. M. M. Knapp, R. B. Yelle, J. J. Juliette and D. R. Tyler, Inorg. Chem., 2009, 48, 7828 Search PubMed; (b) T. J. Sherbow, E. L. Downs, R. I. Sayler, J. J. Razink, J. J. Juliette and D. R. Tyler, ACS Catal., 2014, 4, 3096 Search PubMed; (c) E. L. Downs and D. R. Tyler, J. Inorg. Organomet. Polym., 2014 DOI:10.1007/s10904-014-0079-z , in press.
- (a) R. García-Álvarez, J. Díez, P. Crochet and V. Cadierno, Organometallics, 2010, 29, 3955 Search PubMed; (b) R. García-Álvarez, J. Francos, P. Crochet and V. Cadierno, Tetrahedron Lett., 2011, 52, 4218 Search PubMed; (c) S. E. García-Garrido, J. Francos, V. Cadierno, J.-M. Basset and V. Polshettiwar, ChemSusChem, 2011, 4, 104 Search PubMed; (d) M. L. Buil, V. Cadierno, M. A. Esteruelas, J. Gimeno, J. Herrero, S. Izquierdo and E. Oñate, Organometallics, 2012, 31, 6861 Search PubMed; (e) R. García-Álvarez, S. E. García-Garrido, J. Díez, P. Crochet and V. Cadierno, Eur. J. Inorg. Chem., 2012, 4218 Search PubMed.
- (a) T. J. Geldbach, D. Drago and P. S. Pregosin, Chem. Commun., 2000, 1629 Search PubMed; (b) C. J. den Reijer, M. Wörle and P. S. Pregosin, Organometallics, 2000, 19, 309 Search PubMed; (c) T. J. Geldbach, F. Breher, V. Gramlich, P. G. A. Kumar and P. S. Pregosin, Inorg. Chem., 2004, 43, 1920 Search PubMed.
- (a) S. M. Ashraf, I. Berger, A. A. Nazarov, C. G. Hartinger, M. P. Koroteev, E. E. Nifant'ev and B. K. Keppler, Chem. Biodiversity, 2008, 5, 1640 Search PubMed; (b) S. M. Ashraf, W. Kandioller, M.-G. Mendoza-Ferri, A. A. Nazarov, C. G. Hartinger and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2060 Search PubMed; (c) A. Cavarzan, A. Scarso and G. Strukul, Green Chem., 2010, 12, 790 Search PubMed; (d) H. B. Ammar, X. Miao, C. Fischmeister, L. Toupet and P. H. Dixneuf, Organometallics, 2010, 29, 4234 Search PubMed; (e) S. Kamezaki, S. Akiyama, Y. Kayaki, S. Kuwata and T. Ikariya, Tetrahedron: Asymmetry, 2010, 21, 1169 Search PubMed; (f) E. Bolyog-Nagy, A. Udvardy, F. Joó and A. Kathó, Tetrahedron Lett., 2014, 55, 3615 Search PubMed.
- T. J. Geldbach, D. Drago and P. S. Pregosin, J. Organomet. Chem., 2002, 643–644, 214 Search PubMed.
- G. A. Lawrance, Chem. Rev., 1986, 86, 17 Search PubMed.
- It is well documented that, while the dissociation of one of the chloride ligands in complexes [RuCl2(η6-arene)(PR3)] readily takes place in polar media, the generation of coordinatively unsaturated [Ru(η6-arene)(PR3)2]2+ dications from compounds [RuCl(η6-arene)(PR3)2]+ generally requires their treatment with a chloride abstractor. See, for example: (a) H. Le Bozec, D. Touchard and P. H. Dixneuf, Adv. Organomet. Chem., 1989, 29, 163 Search PubMed; (b) M. A. Bennett, in Comprehensive Organometallic Chemistry II, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon Press, Oxford, 1995, vol. 7, pp. 549–602 Search PubMed; (c) M. A. Bennett, Coord. Chem. Rev., 1997, 166, 225 Search PubMed; (d) J. Gimeno, V. Cadierno and P. Crochet, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2007, vol. 6, pp. 465–550 Search PubMed; (e) J. Gimeno and V. Cadierno, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2007, vol. 6, pp. 551–628 Search PubMed.
- Organosulfur compounds are well-known poisons for homogeneous catalysts due to the formation of relatively strong metal–sulfur bonds: (a) J. Dunleavy, Platinum Met. Rev., 2006, 50, 110 Search PubMed; (b) For an example of the poisoning of a ruthenium catalyst by the methylsulfanyl group, see: A. E. Díaz-Álvarez, P. Crochet and V. Cadierno, Tetrahedron, 2012, 68, 2611 Search PubMed.
- (a) R. S. Ramón, N. Marion and S. P. Nolan, Chem.–Eur. J., 2009, 15, 8695 Search PubMed; (b) G. K. S. Prakash, S. B. Munoz, A. Papp, K. Masood, I. Bychinskaya, T. Mathew and G. A. Olah, Asian J. Org. Chem., 2012, 1, 146 Search PubMed.
- See, for example: (a) S. Park, Y. Choi, H. Han, S. H. Yang and S. Chang, Chem. Commun., 2003, 1936 Search PubMed; (b) H. Fujiwara, Y. Ogasawara, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 5202 Search PubMed; (c) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 3599 Search PubMed; (d) D. Gnanamgari and R. H. Crabtree, Organometallics, 2009, 28, 922 Search PubMed; (e) R. S. Ramón, J. Bosson, S. Díez-González, N. Marion and S. P. Nolan, J. Org. Chem., 2010, 75, 1197 Search PubMed; (f) C. L. Allen, R. Lawrence, L. Emmett and J. M. J. Williams, Adv. Synth. Catal., 2011, 353, 3262 Search PubMed; (g) S. K. Sharma, S. D. Bishopp, C. L. Allen, R. Lawrence, M. J. Bamford, A. A. Lapkin, P. Plucinski, R. J. Watson and J. M. J. Williams, Tetrahedron Lett., 2011, 52, 4252 Search PubMed; (h) P. Kumar, A. K. Singh, R. Pandey and D. S. Pandey, J. Organomet. Chem., 2011, 696, 3454 Search PubMed; (i) R. García-Álvarez, A. E. Díaz-Álvarez, J. Borge, P. Crochet and V. Cadierno, Organometallics, 2012, 31, 6482 Search PubMed; (j) C. Sun, P. Qu and F. Li, Catal. Sci. Technol., 2014, 4, 988 Search PubMed; (k) K. Tambara and G. D. Pantoş, Org. Biomol. Chem., 2013, 11, 2466 Search PubMed.
- (a) F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Dalton Trans., 1987, S1 Search PubMed; (b) A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1 Search PubMed.
- Ruthenium–carbon bond distances in Ru(II)–olefin complexes typically fall within the range 2.15–2.25 Å. See, for example: (a) J. W. Faller and J. K. Chase, Organometallics, 1995, 14, 1592 Search PubMed; (b) J. W. Steed, D. A Tocher and R. D. Rogers, Chem. Commun., 1996, 1598 Search PubMed; (c) N. Feiken, P. S. Pregosin, G. Trabesinger, A. Albinati and G. L. Evoli, Organometallics, 1997, 16, 5756 Search PubMed; (d) E. Lindner, S. Pautz, R. Fawzi and M. Steimann, Organometallics, 1998, 17, 3006 Search PubMed; (e) C. J. den Reijer, D. Drago and P. S. Pregosin, Organometallics, 2001, 20, 2982 Search PubMed; (f) K. Umezawa-Vizzini and T. R. Lee, Organometallics, 2004, 23, 1448 Search PubMed; (g) D. R. Anderson, D. J. O'Leary and R. H. Grubbs, Chem.–Eur. J., 2008, 14, 7536 Search PubMed.
- M. A. Bennett, T.-N. Huang, T. W. Matheson and A. K. Smith, Inorg. Synth., 1982, 21, 74 Search PubMed.
- A. Camp, C. P. Healy, I. D. Jenkins, B. W. Skelton and A. H. White, J. Chem. Soc., Perkin Trans. 1, 1991, 1323 Search PubMed.
- CrysAlisPro CCD & CrysAlisPro RED, Oxford Diffraction Ltd., Oxford, UK, 2008 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 Search PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381 Search PubMed.
- G. M. Sheldrick, SHELXL97: Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- International Tables for X-Ray Crystallography, Kynoch Press, Birminghan, UK, 1974, vol. IV, (present distributor: Kluwer Academic Publishers, Dordrecht, The Netherlands) Search PubMed.
- M. Nardelli, Comput. Chem., 1983, 7, 95 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the reactivity of complex 6 towards water and copies of the NMR spectra of complexes 2b and 7, and the amides synthesized in this work. CCDC 1028020. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra12013b |
| This journal is © The Royal Society of Chemistry 2014 |