
Bio-inspired polydopamine-coated clay and its thermo-oxidative stabilization mechanism for styrene butadiene rubber
Lu Wanga,
Linjia Hub,
Shangbing Gaoa,
Detao Zhaob,
Liqun Zhang*a and
Wencai Wang*b
aKey Laboratory of Beijing City on Preparation and Processing of Novel Polymer Materials, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: zhanglq@mail.buct.edu.cn; Fax: +86-10-64433964; Tel: +86-10-64434860
bState Key Laboratory of Organic-Inorganic Composites, Beijing 100029, China. E-mail: wangw@mail.buct.edu.cn
First published on 18th December 2014
Abstract
Polydopamine (PDA) is labeled as one category of synthetic melanin because it mimics the intriguing radical-scavenging behaviors of its natural counterpart. In this study, PDA modified montmorillonite (PDA-MMT) is utilized as a thermo-oxidative stabilizer for styrene butadiene rubber (SBR). PDA-MMT is fabricated by an aqueous dip-coating, based on the spontaneous alkaline auto-oxidative polymerization of dopamine hydrochloride in an air atmosphere, and is then associated with the SBR matrix via latex compounding. The PDA coating plays an excellent role as a radical-scavenger, and the uniformly dispersed PDA-MMT significantly functions as a physical barrier, which collaboratively work to reduce thermo-induced radical production during the decomposition process of SBR. This mechanism was attested by an in situ thermo-oxidative ageing test along with electron spin resonance (ESR) analysis. Thermal kinetics calculations showing that the apparent activation energy (Ea) of the SBR compounds is augmented by a large margin in the presence of PDA-MMT also corroborate this trend. Above all, the bio-inspired PDA-coating combined with the homogeneous dispersion of MMT exerts a synergistic effect on the thermo-oxidative stabilization of SBR matrix.
1. Introduction
In the recent decades, there has been huge interest in layered silicates (e.g., MMT) acting as an effective nanofiller for polymeric materials,1 as they give an excellent enhancement to the mechanical behaviors2–4 and barrier properties5–8 of polymer/layered silicate composites (PLS), due to their high stiffness, large diameter-thickness ratio, and huge specific surface area. Since the amount of enhancement in such properties is essentially related to the dispersion state and the interfacial interaction of the clay layers, surface modification of the clay is an integral part of the fabrication of PLS.9 This can be carried out via conventional ion exchange reactions, sol–gel linking, atom transfer radical polymerization (ATRP), and polymer intercalation.1Polydopamine constitutes a fascinating class of bio-mimetic polymer inspired by marine mussel adhesion proteins (MAPs)10,11 and has been demonstrated in literature to possess a versatile adhesion to universal surfaces, owing to the strong hydrogen bonding from its abundant catechol groups.9–13
PDA has been the subject of tremendous research effort associated with surface modification14,15 and functionalization.16,17 For instance, our group has manipulated the electroless plating of a silver layer onto various substrates (containing polymeric nanoparticles,18 electrospun-fibers,19 and magnetic metal nanoparticles20) assisted by PDA pre-deposition, as well as by its metal-ion chelating ability.
PDA is also a popular modifier for PLS nanocomposites. Yang and Phua et al. introduced PDA-MMT into epoxy resin and polyether polyurethane, respectively, and thereby obtained the expectant promoted mechanical properties.9,21–23
It is worth noting that PDA is a somewhat melanin-like substance.10 Natural melanin existing in skin, hair, eyes, and other tissues has a photo-protective function.22,24,25 Melanin is able to dissipate absorbed UV radiation into harmless heat and to scavenge some detrimental species, including free radicals, reactive redox metal-ions, and oxidizing species, which can disrupt the normal metabolism in the body.25,26 Although the pathway by which such dissipation functions is still unknown,24 these protective mechanisms are mainly relevant due to their pronounced inhibition capability against harmful radicals.22,27 Since natural melanin is difficult to systematically characterize, due to its structural heterogeneity and chemical complexity,24–26 a synthetic analog originating from dopamine (3,4-dihydroxy-phenylethylamine)28 or L-DOPA (3,4-dihydroxy-phenylalanine)25,29 has so far been used instead as a common substitute in research. In contrast to natural melanin, the synthetic substitute exhibits a comparable or even superior radical-scavenging ability.25,28
It is widely recognized that the ageing mechanisms of polymeric materials seem to be relevant to either thermo-degradation or photolysis, in terms of molecular chains scission as well as cross-linking initiated by reactive free radicals. Therefore some researchers have employed melanin to serve as a novel anti-ageing additive for polymers. Shanmuganathan et al. applied natural melanin extracted from Sepia Officinalis Ink and synthetic melanin derived from L-DOPA to the PMMA matrix, and achieved a notably increased onset of the decomposition temperature of the composite.25
Furthermore, PDA-modified MMT can also be used to develop the anti-ageing ability of PLS nanocomposites. Phua et al. acquired a greatly enhanced UV-resistance of PP/clay composites with the addition of PDA-MMT.22 However, to the best of the authors' knowledge, the performance related to elastomeric materials has yet not been studied.
Toward this end, herein, we try to establish a structure–property–function relationship in SBR-based compounds, and to ascertain the thermo-oxidative stabilization mechanism of PDA-MMT by means of various characterizations. The fact that the PDA coating forms strong interfacial interactions and facilitates the uniform-dispersion of MMT is verified in terms of our optical observations (SEM and TEM). The excellent radical-scavenging ability of the PDA coating alone is characterized by a DPPH Assay (UV-vis). The eminent inhibition effect of thermo-induced carbon-centered radicals is then predicated based on the ESR results. Ultimately, the superior thermo-oxidative stability of SBR/PDA-MMT is evidenced by its accelerated activation energy and the decomposition temperature (TGA), as well as the decrease of unsaturated carbonyl compounds in heat treatment (as assessed by TG-IR).
2. Experimental
2.1 Materials
Sodium montmorillonite (MMT) was supplied by Siping Liufangzi Aska Bentonite Co., Ltd, Jilin Province, China. Emulsion-polymerized styrene butadiene rubber latex (ESBR 1502, solid content = 18.79 wt%) was purchased from Jilin Petrochemical Corporation, Jilin Province, China. Dopamine hydrochloride (Dopamine, 99%), tris(hydroxy-methyl)aminomethane (Tris, 99%), and 2,2-diphenyl-1-picrylhydrazyl (DPPH, 99%) were purchased from Alfar Aser as received. Ethanol and sulphuric acid were obtained from Beijing Chemical Plant, China. All the other ingredients for the rubber mix process were commercial products.2.2 Synthesis of PDA-MMT
Typically, montmorillonite (75 g) was stirred in DI water (1500 mL) for 8 hours (1600 rpm) and rest for at least 24 hours to obtain the supernatant suspension with a concentration of 2 wt% approximately. Then, the clay suspension was exposed to ultrasonic treatment (800 W) for 15 minutes. Dopamine (1.5 g L−1) and Tris (1.2 g L−1) were added into the as-received clay suspension, followed by continuous agitating for 4 hours at ambient temperature. The resulting PDA-MMT was used as a sludge for the later compounding with SBR latex.2.3 Preparation of SBR/PDA-MMT nanocomposites
The SBR/clay nanocomposites were prepared by latex compounding. A pre-determined amount of PDA-MMT suspension was blended with the SBR latex and stirred for 30 minutes. Then, sulphuric acid solution (3 wt%) was incorporated to flocculate the SBR/PDA-MMT rubber particles. The solid products were rinsed until their surface pH appeared to be neutral and then dried for 24 hours at 50 °C. Sequentially, the resulting dried composites proceeded via a common mechanical mixing process. The ingredients used are listed in Table 1. The SBR containing 5 phr PDA-MMT is abbreviated as SBR/PDA-MMT-5 and the same as below.| Ingredient | Loadinga (phr) |
|---|---|
| a Weight parts per 100 weight parts of rubber.b N-Isopropyl-N′-methylphenyl-p-phenylene diamine.c Dibenzothiazole disulfide.d Diphenyl guanidine.e Tetramethyl thiuram disulfide. | |
| SBR | 100 |
| PDA-MMT | 0/5/10/15 |
| Zinc oxide | 5 |
| Stearic acid | 5 |
| Anti-oxidant 4010NAb | 1 |
| Accelerator DMc | 0.5 |
| Accelerator Dd | 0.5 |
| Accelerator TTe | 0.2 |
| Sulfur | 2 |
The SBR/PDA-MMT flocculate rubber was put into a 6 inch double-roll open mill and masticated for 1 minute. Then, zinc oxide, stearic acid, and the anti-oxidant were mixed in turn. Afterwards, the accelerators and sulfur were mingled with the rubber mix. After resting for 24 hours to assure a good diffusion of these ingredients in the matrix, the gross rubber was vulcanized at 143 °C for its corresponding T90 (the optimum cure time). In addition, the control samples of SBR and SBR/MMT were prepared under the same conditions. The final rubber sheets had a thickness ranging from 1.95 mm to 2.05 mm.
2.4 Characterization
The X-ray Photoelectron Spectroscopy (XPS) measurements were carried out on an ESCALAB 250 XPS system (Thermo, USA) with an Al Kα X-ray source (1486.6 eV photons). The X-ray source was run at a reduced power of 150 W, and the pressure of the analysis chamber was constrained at 10−8 torr or even lower. All the binding energies (BEs) were referenced by the C 1s hydrocarbon peak at 284.6 eV to compensate for the surface charging effects. In the peak synthesis, the line width (full width at half maximum (FWHM)) of the Gaussian peaks was kept constant for all the components in a particular spectrum.The smooth rubber sheets (with sizes of about 10 mm × 10 mm × 2 mm) were scanned from 0.5° to 10° at a scanning rate of 1° min−1 using D/Max2500 VB2+/PC X-ray Diffraction (Rigaku, Japan) with Cu Kα radiation. The TEM observations were conducted on an H-800 Transmission Electron Microscope (HITACHI, Japan) at 200 kV. The ultrathin sections of SBR/clay nanocompounds for TEM were cut using a microtome (LEICA EM FC7). The Scanning Electron Microscope images were captured by a S-4800 SEM (HITACHI, Japan) at an accelerating voltage of 20 kV. The rubber samples were broken off in liquid nitrogen, and the flat worn fractures were selected for SEM. In addition, a thin layer of platinum was sputtered on the sample surfaces prior to the SEM observations.
The mechanical properties were performed by means of a universe electronic testing machine (SANS CMT 4104, China) according to the National Standards of China or ASTM with a tensile speed of 500 mm min−1 at ambient temperature. The rubber sheets were die-cut into dumbbell-shaped samples with a working-district width of 6 mm. For each group of data reported, at least five sample measurements were taken and averaged.
The radical scavenging property of PDA was determined by a DPPH Spectrophotometry Assay22,30 with a slight alteration along with a UV-3600 UV-vis spectrometer (Shimadzu, Japan). The 0.05 mg mL−1 solution of DPPH in ethanol was freshly prepared, and 500 μL of pristine PDA aqueous solution (dopamine 1.5 g L−1, pH = 8.5, 4 h) was diluted with 14 mL of DI water prior to usage. The scavenging activity was characterized by monitoring the decrease in the UV-vis absorption of DPPH at 517 nm at different dose levels of PDA (the addition amounts of diluted PDA solution varied from 100 μL to 800 μL). To enable an impartial comparison, ascorbic acid was chosen as a positive control. The calculation equation for the radical-inhibition capability is as follows:
| Inhibition = [1 − (Ai − Aj)/A0]. |
The ESR apparatus used here was a JES-FA 200 X-band Electron Spin Resonance spectrometer (JEOL, Japan) with a temperature accessory (JEOL, CVT Controller). The rod-shaped sample was stored in a spectrosil tube (3.8 mm i.d.) in air atmosphere and measured at a 100 kHz modulation frequency with a microwave power of 0.1 mW. The center magnetic field was 3234.91 Gauss with a sweep width of (±) 75 Gauss. The standard sample of manganese (Mn2+) inserted beside the specimen was applied to collimate the center field and the magnetic field strength. The SBR/clay samples were aged using the temperature accessory at 150 °C for various times, at which point the resonance signals were recorded accurately by ESR.
The SBR/clay samples were heated from 40 °C to 600 °C at a heating rate of 10 °C min−1 in air atmosphere with a purge rate of 50 mL min−1 by means of Thermo-Gravimetric Analysis (METTLER-TOLEDO, Switzerland). To ascertain the apparent activation energy (Ea), the samples were heated at different heating rates of 5 °C min−1, 10 °C min−1, 20 °C min−1 and 30 °C min−1, respectively. Ea was calculated according to the classical Flynn–Wall–Ozawa method.31,32 Additionally, Thermo-Gravimetry coupled to Fourier Transform Infrared spectroscopy was conducted to investigate the gaseous products of the specimens evolved during the thermal treatment. TG measurements were performed as described above (heating from 40 °C to ∼700 °C). Infrared spectra were recorded using a Nicolet 6700 FT-IR (Thermo, USA) equipped with a DLaTGS detector in the 400–4000 cm−1 range with 4 cm−1 spectral resolution over 32 scans. The temperature of the transfer line linking TG and FT-IR was 215 °C.
3. Results and discussion
3.1 Surface modification of MMT by PDA
Although the polymerization mechanism of dopamine has been reported extensively, it is still under some debate. It is widely accepted that the catechol group of dopamine is easily oxidized to quinone under an alkaline aqueous environment.9,10,28 After a set of oxidation, cyclization, and rearrangement reactions,33 a key metastable intermediate—5,6-dihydroxy-indole (DHI)26—emerges and is able to react with its oxidized form (IQ)26 to generate semi-quinone radicals (SQ)26 via dismutation33 (see Scheme 1). Then, dopamine polymerization progresses through a radical-induced intra-molecular cross-linking pathway. | ||
| Scheme 1 Proposed mechanism for the polymerization of dopamine (a-oxidation, b-cyclization, c-rearrangement, and d-dismutation). | ||
XPS is used to probe the chemical composition and corresponding bonding status of MMT and PDA-MMT. After being coated by PDA, the signal of the characteristic nitrogen is detectable at binding energy (BE) of 399.75 eV from the wide scan spectra of XPS (see Fig. 1(a) and (b)). In Fig. 1(c), the C 1s core-level spectrum of unmodified MMT can be curved-fitted with two peak components, with BE at 282.5 eV for the C–Si impure species, and at 284.6 eV for the C–H species (carbon dioxide contamination). The C 1s core-level spectrum of PDA-MMT can be curved-fitted with three peak components in Fig. 1(d), with BE at 284.6 eV for the C–H species, 285.5 eV for the C–N species, and 287.5 for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species. In Fig. 1(f), the N 1s core-level spectrum of PDA-MMT comprises two peak components at 388.5 eV for –N, and 389.5 eV for –N–H. The CO species from the quinone derivatives and the –N species from the indole compounds illustrate the formation of polydopamine deposited onto MMT, which consolidates the proposed polymerization mechanism of dopamine as mentioned above.
O species. In Fig. 1(f), the N 1s core-level spectrum of PDA-MMT comprises two peak components at 388.5 eV for –N, and 389.5 eV for –N–H. The CO species from the quinone derivatives and the –N species from the indole compounds illustrate the formation of polydopamine deposited onto MMT, which consolidates the proposed polymerization mechanism of dopamine as mentioned above.
 | ||
| Fig. 1 XPS wide-scan spectra, C 1s core-level spectra and N 1s core-level spectra of (a, c and e) MMT and (b, d and f) PDA-MMT. | ||
The TEM images are exploited here to produce direct evidence of the morphological alterations of MMT before and after the PDA surface modifications (see Fig. 2(a) and (b)). As PDA exerts an influence of adhesion rather than intercalation to some degree,9,21 ultrasonic treatment is carried preceding the deposition of PDA, to decrease the aggregation of MMT. In Fig. 2(a) and (b), after ultrasonic treatment, most MMT is in the form of a mono-layer distribution, with a width ranging from 200 nm to 1000 nm, whereas PDA-MMT is still dispersed arbitrarily on the TEM grids in similar dimensions.
 | ||
| Fig. 2 TEM images of (a) MMT, (b) PDA-MMT, (c and d) SBR/MMT-5, and (e and f) SBR/PDA-MMT-5. | ||
3.2 Morphology and mechanical properties of the SBR/PDA-MMT nanocomposites
The XRD patterns of pristine MMT, SBR, SBR/MMT, and SBR/PDA-MMT are shown in Fig. 3. Firstly, it can be seen that the pristine MMT (without ultrasonic treatment) has a feature peak at approximately 6°. This peak weakens greatly when MMT (ultrasonic) is compounded with the SBR matrix, whereas it is extinguished with PDA coating. The ultrasonic treatments combined with the PDA coating improve the good dispersion of clay in the matrix. On the other hand, SBR chains intercalation into the clay galleries displays a feature peak at lower degrees (2θ ≈ 2°), corresponding to the larger spatial distance.4 Similarly, the intensity of such intercalation peak is lessened substantially, demonstrating the destroyed micro-structure of either ultrasonic MMT or PDA-MMT. Moreover, the intercalation intensity of SBR/PDA-MMT is by far lower than that of SBR/MMT, indicating a uniform dispersion of PDA-MMT layers, instead of the relatively intense aggregation in the matrix. With the increase in clay content, those peaks gradually intensify, symbolizing the inevitable re-aggregation of clay. We hypothesize that this phenomenon mainly arises from the mechanical shear during the mixing and heat-pressing in the vulcanization process. Nevertheless, the intensity of such peaks in SBR/MMT always exceeds that of SBR/PDA-MMT at the same clay loading level, implying that the strong interactions between PDA and SBR defy the re-aggregation of clay. Thus, PDA-MMT is considered homogeneously dispersed in the SBR matrix in terms of the nearly flat XRD patterns, that is to say, the X-ray diffraction from the distributed silicate layers have mostly disappeared.4 | ||
| Fig. 3 XRD patterns of pristine MMT, SBR and SBR/clay nanocomposites. | ||
Since XRD is not a quantitative analysis in which PDA-MMT might be easily diluted by the SBR matrix, conclusions regarding the spatial distribution based on XRD alone are not adequate. Related TEM images are captured in Fig. 2(c)–(f). A much rougher and thicker agglomeration of MMT (500–1000 nm) is observed in SBR/MMT, indicating an inferior dispersion, namely an intercalated and flocculated dispersion.4 Paradoxically, uniformly-intercalated PDA-MMT layers can be scarcely discerned from the low contrast background (as shown in Fig. 2(e), in which the black dots should relate to some ingredients in the SBR/clay composite). In amplifying the magnification in Fig. 2(f), PDA-MMT is in the form of a slim laminate (200–500 nm), which assumes a light-fog appearance.
SEM observations of the worn fractures of the SBR/clay nanocompounds were performed to illuminate the interfacial interactions between the filler and matrix. The sparkling spots are thought to be some conductive additive (e.g., zinc oxide) added during the mix process. Defects or holes can be barely detected in pristine SBR, while the vicinity of MMT brings in many defects in to the matrix (see Fig. 4(a) and (b)). As shown in Fig. 4(c), with the addition of PDA-MMT, the shortcomings vanish, and the boundaries between clay and SBR blur. In fine detail, the strong hydrogen bonding from the abundant catechol groups of PDA and the π–π conjugation of the pendant aromatic structures give rise to impressive interfacial interactions.
 | ||
| Fig. 4 SEM images of (a) SBR, (b) SBR/MMT-5, and (c) SBR/PDA-MMT-5. | ||
The tensile properties of SBR/clay nanocompounds are shown in Table 2. The tensile strength of SBR is only 1.8 MPa, while it is increased by more than 150% through the addition of only 5 phr PDA-MMT, much higher than that of MMT at the same loading level. As the stress gradually improves, the interfacial interaction associated with the cross-linking network begins to take power.34 The modulus at 300% is increased to 2.6 MPa in SBR/PDA-MMT at a very low loading content (5 phr), implying that PDA-MMT possesses robust interfacial interactions with SBR, which thus function as efficient physical cross-linkers.9,34 Namely, the PDA coating is able to lubricate the interface during the tensile process, which diminishes the slippage and fracture of SBR chains anchored onto the MMT galleries, which favors the stress-average, and thus contributes to the enhancement of tensile strength, as well as to the break elongation.34 However, with the increase in the clay loading, a sustainable improvement of the tensile properties could hardly be found. Moreover, this situation was also found in associated literature.21,22 Besides, as layered silicate is not an optimal reinforcing filler for rubber compared with sphere fillers (e.g., carbon black and silica), the reduction in tensile properties could be partially attributed to the aggregation dispersion of PDA-MMT at higher loading levels.
| Sample | Modulus at 100% (MPa) | Modulus at 200% (MPa) | Modulus at 300% (MPa) | Break elongation (%) | Tensile strength (MPa) |
|---|---|---|---|---|---|
| SBR | 0.8 | 1.1 | 1.3 | 426 | 1.8 |
| SBR/PDA-MMT-5 | 0.9 | 1.5 | 2.6 | 470 | 5.0 |
| SBR/MMT-5 | 1.1 | 1.4 | 1.8 | 438 | 2.4 |
| SBR/PDA-MMT-10 | 1.0 | 1.4 | 1.8 | 669 | 4.3 |
| SBR/MMT-10 | 0.9 | 1.0 | 1.2 | 577 | 2.4 |
| SBR/PDA-MMT-15 | 1.0 | 1.5 | 2.0 | 647 | 4.5 |
| SBR/MMT-15 | 1.0 | 1.4 | 1.7 | 640 | 3.1 |
Nevertheless, the possibility of covalent bonds could not be excluded absolutely,9 as the remaining aminogen (–NH2) from PDA might react with the residual double bonds of the SBR chains.
3.3 Thermo-oxidative stabilization mechanism for SBR/PDA-MMT
As mentioned above, melanin exhibits a unique radical-scavenging ability, and consequently it has been conceived as a free radical trap (also termed as an electron trap), owing to its unique radical character.35–37 Mason et al. noted that both natural and synthetic melanin displayed a sharp ESR absorption, implying the presence of free radicals. Based on the model of synthetic melanin, they attributed this radical property to a stabilized semiquinonoid form.29 Also, Simon ascribed this remarkable property of melanin to its distinct hydroquinone moiety.24 As described in Section 3.1, the formation of PDA has been proven to undergo a radical-induced intra-molecular coupling pathway similar to that of melanin.10,26,33 Furthermore, the important hydroquinone intermediate 5,6-dihydroxy-indole (DHI) and its various oxidized forms are indeed the decisive precursors or building blocks of melanin26 and are responsible for the yield of semiquinone radicals. Thus, the synthetic PDA coating could help SBR to resist thermo-oxidation deterioration by mimicking natural melanin. That is, PDA reacts with reactive radicals via a one-electron donating or radical-addition pathway.25It is rather interesting to note that even dopamine itself has a radical-scavenging activity through a one-step hydrogen atom transfer (HAT) strategy or metal-ion assisted HAT from its catechol groups to reactive oxygen species (ROS).38 In that case, dopamine itself (as one kind of neurotransmitter) could quench these ROS in neurotransmission systems. However, it is difficult to see how the operation of such a mechanism could come about in PDA, due to the absolutely different chemical structure between the monomer and the corresponding macromolecule.
Since radical-induced chain scission is a common thermo-oxidative mechanism for most polymers,25 it is of great significance in our present work to scrutinize whether PDA-MMT could be employed as an efficient anti-aging additive for SBR.
After the addition of PDA aqueous solution, the dark purple solution of DPPH converted to a yellow-colored one, because of reduction of the stable DPPH radical. Fig. 5(a) shows the decay of the UV-vis absorbance spectra at 517 nm upon the addition of PDA. Fig. 5(b) illustrates the corresponding scavenging activity of PDA compared with the reference ascorbic acid (an effective radical-scavenging anti-oxidant) at the same aliquot level. Both reveal a similar tendency of scavenging activity. The scavenging activity dose-dependently ascends rapidly during the early stage and shows a quasi-linear trend. Eventually, the scavenging effect of PDA, as well as ascorbic acid, reaches the maximum value of 85.1% and 95.6%, respectively. These results signify that the PDA coating has an impressive scavenging capability, which is comparable with ascorbic acid.
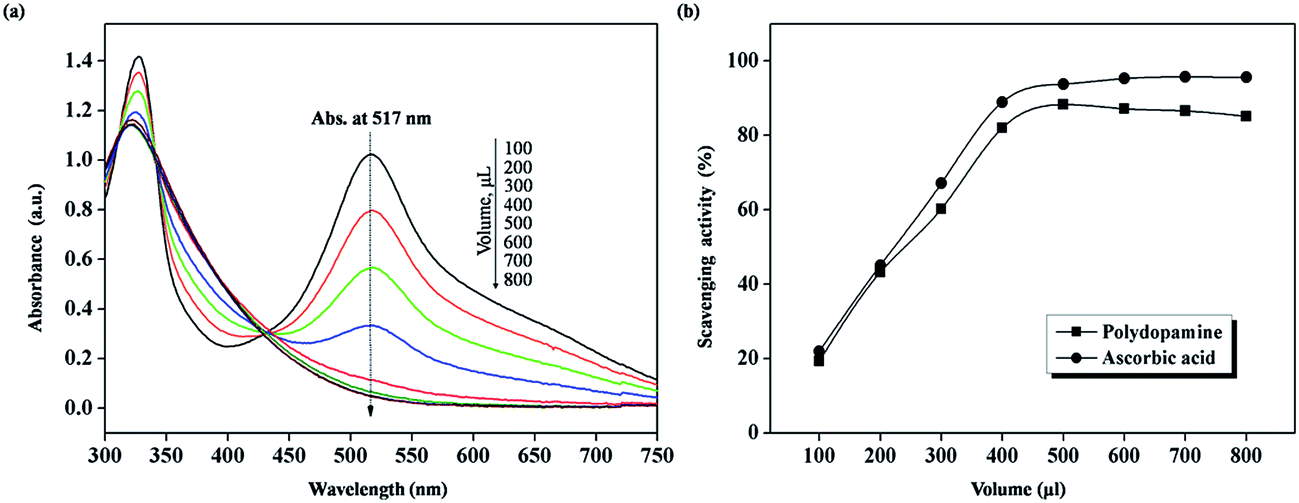 | ||
| Fig. 5 (a) UV-vis spectra of DPPH ethanol solution decrease with the addition of PDA solution (5.17 × 10−2 g L−1) at ambient temperature. (b) Scavenging activity of sample (PDA) and control standard (ascorbic acid) on DPPH radicals at various addition volumes (solid square: polydopamine and solid circle: ascorbic acid). | ||
Notwithstanding this, PDA-MMT is also endowed with a higher specific surface area, which means more available reactive sites,22,28 and thus it exhibits an inferior scavenging ability on DPPH radicals by this method. The reason for this could be that PDA-MMT is a sort of suspension rather than a solution, whereas a homogeneous solution is fundamental when applying spectroscopic measurements. Nevertheless, based on the pronounced scavenging activity on DPPH of PDA, it is a reasonable presumption to make that the high-dispersion clay platelets deposited by PDA would impart an excellent inhibition effect to thermo-induced radicals in SBR matrix. Further research related to this matter of PDA-MMT is outlined below.
Elastomeric material is also a major subject for ESR measurement.44–46 ESR measurements allow reasonable mechanism analyses, with most of these measurements achieved below the glass transition temperature of polymer41–44 under which the motion of polymer chains is blocked to a large extent. Herein, it is doubtful whether the practical anti-ageing property of polymeric materials could be determined under such rigid conditions. To circumvent this issue, we established a sort of in situ thermo-oxidative ageing test along with ESR. The evolution of the spectral shape and the relative spin generation efficiency were determined to ascertain the expectant excellent scavenging activity of PDA-MMT.
As shown in Fig. 6(a) and (b), thermo-induced radicals generated in SBR/clay compounds give a broad and para-symmetrical singlet eventually between the two standard Mn2+ peaks. It is known that alkyl radicals are implicated in the thermal degradation of polymer, therefore the authors tentatively assigned these spectra to alkyl radicals (maybe some alkoxyl radicals concerned47). The interpretation that the broad singlet originates from alkyl radicals could be verified by using pristine SBR for ESR testing, which is known to produce such radicals. As shown in Fig. 6(c), the spectrum gives a similar singlet that is even sharper.
 | ||
| Fig. 6 ESR spectra measured at 150 °C at various ageing times of (a) SBR/MMT-10, (b) SBR/PDA-MMT-10, and (c) SBR. (d) The evolution of ESR spectra after ageing for 30 minutes at 170 °C. | ||
The spectral differences between SBR and SBR/clay indeed reveal the outstanding thermo-oxidative stabilization of PDA-MMT. At the early stage of degradation, SBR/PDA-MMT and SBR/MMT exhibit similar spectra shapes. The resonance signals are rather weak, and the features of the central part are masked by a drift of the baseline, which is caused by the high executive temperature (Fig. 6(a) and (b)). The height of the singlet of SBR/MMT increases rapidly as the ageing-times prolong, while the emergence of such a sharp singlet is delayed substantially in the pattern of SBR/PDA-MMT.
It could be inferred that, firstly, high-dispersion clay platelets prevent the intrusion of heating, as well as the diffusion of contagious species, and then the PDA coating handicaps the generation of thermo-induced radicals. In contrast to those without PDA in Fig. 6(d), the characteristic singlet shifts forward. Both the spectral transformation and the aforementioned alterations of the peak shape confirm the occurrence of chemical scavenging on the carbon-centered radicals of the PDA coating.43,47
Moreover, the relative spin generation was calculated by the intensity ratio of the SBR/clay and Mn2+, in which the standard sample Mn2+ was utilized to calibrate for the errors arising from the slight fluctuation of the measuring parameters. The relative spin generation is plotted in Fig. 7 as a function of aging time. The relative spin generation efficiency is obtained from the slope of the plot. SBR/PDA-MMT gives an efficiency value of 0.021 a.u. min−1, which is lower than that of SBR/MMT (0.036 a.u. min−1), indicating a superior inhibition effect on the reactive species of PDA-MMT.
 | ||
| Fig. 7 Relative spin generation as a function of ageing time (solid circle: SBR/MMT-10 and solid square: SBR/PDA-MMT-10). For the calibration of spin generation, the standard sample of manganese (Mn2+) was used. | ||
However, the pivotal reactive peroxyl radicals in thermo-oxidative decomposition could not be detected, due to their short life under high temperature. However, for a full understanding, detailed and more convincing grounds should be acquired in a more rational manner of ESR measurement and combined with other spectroscopic methods.43
 | ||
| Fig. 8 TGA and corresponding DTG curves of SBR/clay nanocomposites in an air atmosphere at a heating rate of 10 °C min−1. | ||
| Sample | Decomposition temperature (°C) | ||||
|---|---|---|---|---|---|
| T10% | T20% | T50% | T70% | Tmax | |
| SBR | 395.3 | 408.5 | 427.8 | 439.0 | 426.0 |
| SBR/MMT | 389.0 | 405.9 | 425.4 | 443.1 | 422.7 |
| SBR/PDA-MMT | 385.3 | 406.3 | 439.0 | 457.7 | 449.5 |
In order to quantitate the thermo-stabilization effect of PDA-MMT, the apparent activation energy of degradation (Ea) with respect to different conversions was investigated by the Flynn–Wall–Ozawa method.25,31,32 The activation energy is calculated from the slope of the linear fitting of the data using the following equation:

The TGA curves of SBR-based compounds at different heating rates are shown in Fig. 9(a)–(c). When the heating rate improves, the corresponding TG-DTG curves shift to higher temperature, due to the thermal hysteresis effect, indicating that at the same weight loss percentage, the decomposition temperature rises with the increasing heat rate.
 | ||
| Fig. 9 TGA and corresponding DTG curves of (a) SBR/MMT-10 and (b) SBR/PDA-MMT-10 (in air atmosphere at different heating rates). | ||
The Ea of SBR-based compounds varies at different conversions, as described in Table 4. It gradually increases until reaching a maximum, implying the random chain scission and the formation of a peroxyl radical intermediate (the latter necessitates more energy). The subsequent decrease of Ea signifies the swift auto-catalyzed oxidizing of the SBR chains. The Ea of SBR/PDA-MMT exceeds that of SBR/MMT, with the amount of enhancement falling within a range of 10–90 kJ mol−1 across the different degrees of conversion. On the other hand, the lower Ea of SBR/MMT can be anticipated according to its lower decomposition temperature aforementioned. Based on the above, PDA-MMT instead of MMT itself exerts a remarkable stabilization effect upon SBR.
| Conversion (%) | Ea (kJ mol−1) | ||
|---|---|---|---|
| SBR | SBR/MMT | SBR/PDA-MMT | |
| 10 | 487.0 | 436.6 | 497.9 |
| 20 | 497.1 | 443.0 | 528.0 |
| 30 | 431.5 | 411.4 | 488.4 |
| 50 | 321.6 | 348.2 | 362.2 |
| 70 | 301.5 | 334.0 | 332.1 |
To gain more evidence, a method to detect the gaseous products of SBR-based compounds evolved during thermal treatment was developed by overlapping a series of temporal profiles of the infrared absorption bands. The main infrared absorption bands of the thermo-oxidation products lie in the range of 1700–1800 cm−1 of unsaturated carbonyl compounds, 2400 cm−1 of carbon dioxide and 3400–3900 cm−1 of water vapor.
Fig. 10 presents the 3D TG-IR images of SBR and SBR/clay nanocomposites. It is observed that the intense infrared absorption mostly occurs in the temperature interval between 390 °C and 600 °C, which is consistent with the TGA results above. As a consequence of the thermo-oxidative decomposition, the characteristic infrared absorption bands of the unsaturated carbonyl compounds are detectable in the range of 1700–1800 cm−1, which consists of carboxyl (1716 cm−1), ketone (1750 cm−1), lactone (1780 cm−1), and so forth. Compared with pristine SBR, the MMT layers alone restrain the SBR matrix from severe decomposition, as is shown in Fig. 10(a) and (b). It is striking to note that the PDA-MMT layers substantially prevent that trend by decreasing the generation of such oxidation products, as seen in Fig. 10(c).
 | ||
| Fig. 10 3D infrared absorption spectra as a function of temperature for the gaseous products of (a) SBR, (b) SBR/MMT-10 and (c) SBR/PDA-MMT-10 that evolve during thermal treatment. | ||
In all, PDA-MMT considerably improves the thermo-oxidative stability of the SBR matrix. Such enhancement could be partially attributed to the strengthened interface, as SBR chains need more energy to defy the steric hindrance of PDA-MMT. However, the predominant factor is that PDA acts as a scavenger of carbon-centered radicals, thus screening the SBR matrix from rapid thermal degradation. Additionally, this polymeric pigment (PDA) has a property of chelating metal-ions.10,16 The thermal stability of the SBR matrix could also be improved in the presence of PDA, in that some residual metal complex may aggravate the decomposition process.
4. Conclusion
PDA-MMT was prepared by a facile dip-coating method. The MMT platelets intercalated uniformly in the SBR matrix with recourse to PDA coating. PDA-MMT significantly improved the tensile strength of SBR by more than 150% at a very low loading content (5 phr). The incorporation of PDA-MMT developed the specific weight loss temperature of SBR, and the maximum weight loss temperature of SBR/PDA-MMT was elevated by 26.8 °C. Moreover, the enhancement value of the decomposition activation energy (Ea) of the nanocompounds ranges from 10–90 kJ mol−1 throughout the different degrees of conversion. Furthermore, PDA-MMT diminishes the amount of the unsaturated carbonyl oxidative products of SBR that evolve during thermal treatment. The PDA coating alone even displays a dramatic DPPH radical-scavenging efficiency (85.1%) that is comparable to that of ascorbic acid (95.1%). PDA-MMT causes a desirable inhibition effect on the thermo-induced carbon-centered radicals of SBR during the course of thermo-oxidative degradation in the ESR testing.In summary, due to the chemical scavenging activity of the PDA coating as well as the physical barrier of the high-dispersion clay layers, PDA-MMT exerts an effective thermo-oxidative stabilization effect on the SBR matrix.
Acknowledgements
The authors sincerely appreciate the financial supports from the Natural Science Foundation of China (Grant no. 51373010, Grant no. 51221002), and the Program for New Century Excellent Talents in University (NCET-11-0562). Meanwhile, the authors wish to express their gratitude to Dr Yang Haijun from Analysis Center of Tsinghua University for his kindly suggestions related to ESR measurements in this study.References
- C. W. Chiu, T. K. Huang, Y. C. Wang, B. G. Alamani and J. J. Lin, Prog. Polym. Sci., 2014, 39, 443–485 CrossRef CAS PubMed.
- A. A. Azeez, K. Y. Rhee, S. J. Park and D. Hui, Composites, Part B, 2013, 45, 308–320 CrossRef CAS PubMed.
- L. Q. Zhang, Y. Z. Wang, Y. Q. Wang, Y. Sui and D. S. Yu, J. Appl. Polym. Sci., 2000, 78, 1873–1878 CrossRef CAS.
- S. Sinha Ray and M. Okamoto, Prog. Polym. Sci., 2003, 28, 1539–1641 CrossRef PubMed.
- J. J. Kochumalayil, M. Bergenstråhle-Wohlert, S. Utsel, L. Wågberg, Q. Zhou and L. A. Berglund, Biomacromolecules, 2013, 14, 84–91 CrossRef CAS PubMed.
- Y. P. Wu, Y. Q. Wang, H. F. Zhang, Y. Z. Wang, D. S. Yu, L. Q. Zhang and J. Yang, Compos. Sci. Technol., 2005, 65, 1195–1202 CrossRef CAS PubMed.
- S. Livi, G. Sar, V. Bugatti, E. Espucheacd and J. Duchet-Rumeau, RSC Adv., 2014, 4, 26452–26461 RSC.
- H. W. P. Carvalho, C. V. Santilli, V. Briois and S. H. Pulcinelli, RSC Adv., 2013, 3, 22830–22833 RSC.
- L. Yang, S. L. Phua, J. K. H. Teo, C. L. Toh, S. K. Lau, J. Ma and X. Lu, ACS Appl. Mater. Interfaces, 2011, 3, 3026–3032 CAS.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- M. S. Ata, Y. Liu and I. Zhitomirsky, RSC Adv., 2014, 4, 22716–22732 RSC.
- Y. Liu and K. Li, Macromol. Rapid Commun., 2002, 23, 739–742 CrossRef CAS.
- M. C. van der Leeden, Langmuir, 2005, 21, 11373–11379 CrossRef CAS PubMed.
- Z. Y. Xi, Y. Y. Xu, L. P. Zhu, Y. Wang and B. K. Zhu, J. Membr. Sci., 2009, 327, 244–253 CrossRef CAS PubMed.
- X. M. Zhang, Z. Y. Li, X. B. Yuan, Z. D. Cui, H. J. Bao, X. Li, Y. D. Liu and X. J. Yang, Mater. Sci. Eng., C, 2013, 33, 2816–2820 CrossRef CAS PubMed.
- Q. Ye, F. Zhou and W. M. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC.
- J. Ryu, S. H. Ku, H. Lee and C. B. Park, Adv. Funct. Mater., 2010, 20, 2132–2139 CrossRef CAS.
- Y. Jiang and W. C. Wang, Polym. Adv. Technol., 2011, 22, 2509–2516 CrossRef CAS.
- Y. Fu, L. Liu, L. Q. Zhang and W. C. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 5105–5112 CAS.
- Q. Li, M. Tian, L. Liu, H. Zou, L. Q. Zhang and W. C. Wang, Electrochim. Acta, 2013, 91, 114–121 CrossRef CAS PubMed.
- S. L. Phua, L. Yang, C. L. Toh, S. Huang, Z. Tsakadze, S. K. Lau, Y. W. Mai and X. Lu, ACS Appl. Mater. Interfaces, 2012, 4, 4571–4578 CAS.
- S. L. Phua, L. Yang, C. L. Toh, D. Guoqiang, S. K. Lau, S. K. Lau, A. Dasari and X. Lu, ACS Appl. Mater. Interfaces, 2013, 5, 1302–1309 CAS.
- S. Huang, S. L. Phua, W. Liu, G. Ding and X. Lu, RSC Adv., 2014, 4, 1425–1431 RSC.
- J. D. Simon, Acc. Chem. Res., 2000, 33, 307–313 CrossRef CAS PubMed.
- K. Shanmuganathan, J. H. Cho, P. Iyer, S. Baranowitz and C. J. Ellison, Macromolecules, 2011, 44, 9499–9507 CrossRef CAS.
- P. Meredith, B. J. Powell, J. Riesz, S. P. Nighswander-Rempel, M. R. Pederson and E. G. Moore, Soft Matter, 2006, 2, 37–44 RSC.
- F. Daniels, J. Invest. Dermatol., 1959, 32, 147–155 CrossRef.
- K. Y. Ju, Y. Lee, S. Lee, S. B. Park and J. K. Lee, Biomacromolecules, 2011, 12, 625–632 CrossRef CAS PubMed.
- H. S. Mason, D. J. E. Ingram and B. Allen, Arch. Biochem. Biophys., 1960, 86, 225–230 CrossRef CAS.
- Y. Chen, M. Y. Xie, S. P. Nie, C. Li and Y. X. Wang, Food Chem., 2008, 107, 231–241 CrossRef CAS PubMed.
- J. H. Flynn and L. A. Wall, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 323–328 CrossRef CAS.
- T. Ozawa, Bull. Chem. Soc. Jpn., 1965, 38, 1881–1886 CrossRef CAS.
- W. C. Wang, R. Y. Li, M. Tian, L. Liu, H. Zou, X. Y. Zhao and L. Q. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 2062–2069 CAS.
- Z. H. Wang, J. Liu, S. Z. Wu, W. C. Wang and L. Q. Zhang, Phys. Chem. Chem. Phys., 2010, 12, 3014–3030 RSC.
- C. C. Felix, J. S. Hyde, T. Sarna and R. C. Sealy, J. Am. Chem. Soc., 1978, 100, 3922–3926 CrossRef CAS.
- B. Commoner, J. Townsend and G. E. Pake, Nature, 1954, 174, 689 CrossRef CAS.
- R. Dunford, E. J. Land, M. Rozanowska, T. Sarna and T. G. Truscott, Free Radical Biol. Med., 1995, 19, 735–740 CrossRef CAS.
- T. Kawashima, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. B, 2009, 114, 675–680 CrossRef PubMed.
- Advanced ESR Methods in Polymer Research, ed. S. Schlick, John Wiley & Sons, 2006, pp. 102–105 Search PubMed.
- B. Ellis and J. F. Baugher, J. Polym. Sci., Polym. Phys. Ed., 1973, 11, 1461–1463 CAS.
- M. Sakaguchi, S. Kodama, O. Edlund and J. Sohma, J. Polym. Sci., Polym. Lett. Ed., 1974, 12, 609–613 CrossRef CAS.
- Z. Osawa, E. L. Cheu and Y. Ogiwara, J. Polym. Sci., Polym. Lett. Ed., 1975, 13, 535–542 CrossRef CAS.
- A. Tkáč and I. Špilda, J. Polym. Sci., Polym. Chem. Ed., 1981, 19, 1495–1508 CrossRef.
- J. Čulin, D. Gembarovski, M. Andreis, Z. Veksli and T. Marinović, Polym. Int., 2000, 49, 845–852 CrossRef.
- N. Suzuki, M. Ito and F. Yatsuyanagi, Polymer, 2005, 46, 193–201 CrossRef CAS PubMed.
- M. Ito, H. Isago and N. Suzuki, J. Appl. Polym. Sci., 2008, 108, 1385–1392 CrossRef CAS.
- T. Takeshita, K. Tsuji and T. Seiki, J. Polym. Sci., Polym. Chem. Ed., 1972, 10, 2315–2324 CAS.
| This journal is © The Royal Society of Chemistry 2015 |