
One pot microwave assisted synthesis of bisphosphonate alkene capped gold nanoparticles†
Romain Aufaurea,
Yoann Lalatonne*ab,
Nicole Lièvrec,
Olivier Heintzd,
Laurence Mottea and
Erwann Guénin*a
aUniversité Paris 13, Sorbonne Paris Cité, Laboratoire CSPBAT, CNRS (UMR 7244), 74 avenue M. Cachin, 93017 Bobigny, France. E-mail: guenin@univ-paris13.fr; yoann.lalatonne@avc.aphp.fr; Fax: +33 141088528; Tel: +33 148387621
bDepartment of Nuclear Medecine, Avicenne Hospital, Université Paris 13, Sorbonne Paris Cité, 125 rue de Stalingrad, 93009 Bobigny, France
cUniversité Paris 13, Sorbonne Paris Cité, UPRES 3410 Biothérapies Bénéfices et Risques, CNRS (UMR 7244), 74 avenue M. Cachin, 93017 Bobigny, France
dUniversité de Bourgogne, Laboratoire Interdisciplinaire Carnot de Bourgogne, CNRS (UMR 5209), 9 Avenue Alain Savary, BP 47870, 21078 Dijon Cedex, France
First published on 3rd November 2014
Abstract
A new synthetic pathway for the direct synthesis of water soluble gold nanoparticles (GNPs) already possessing terminal alkene functional groups was developed. This is achieved by using synthesized (1-hydroxy-1-phosphonopent-4-enyl)phosphonic acid (HMBPene), presenting advantages of the well known bisphosphonate coating applied to colloidal gold instead of metal oxides. The proposed protocol allowed an accurate control of the particle size in the 13–20 nm diameter range with a high spherical uniformity, which is a crucial point for these colloids' properties. We have shown that it is possible to synthesize a functionalized nanoplatform in a one-pot one-phase process with a sacrificial molecule as reductant, pH mediator and capping agent.
Introduction
GNPs have become one of the most commonly studied metallic colloids for a variety of potential applications in catalysis, biology, and optics due to the Surface Plasmon Resonance (SPR) properties.1–6 This phenomenon strongly depends of the particle size & shape, inter-particle distance, and the nature of the capping organic shell. Consequently, the control of the crystalline growth and functionalization are crucial challenges in the development of GNPs synthesis. Hence a comprehension of Au(III) reduction and nanocrystal nucleation-growth mechanism is of critical importance. In aqueous media, the citrate reduction of gold described by Turkevich in 1951![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 and improved by Frens in 19738 is an easy reproducible technique still widely use today. Several studies have been done on the overall reaction mechanism9–13 leading to different interpretations. Nevertheless some common points can be established: gold ions are firstly reduced to form nuclei (around 2 nm), which self-assemble in seeds by Ostwald ripening and finally the crystalline core growth occur with remaining solvated ions. Moreover, Ji et al. has proposed a pH-dependant model to streamline the nanospheres final diameter according to UV-visible records and TEM images of intermediates states.13 Two different reaction pathways were identified. The first one that occurred for the acidic pH range in a three steps process: fast nucleation, random attachment to nanowires, and intra-particle ripening. The second pathway under high pH is in line with the nucleation-growth model.
7 and improved by Frens in 19738 is an easy reproducible technique still widely use today. Several studies have been done on the overall reaction mechanism9–13 leading to different interpretations. Nevertheless some common points can be established: gold ions are firstly reduced to form nuclei (around 2 nm), which self-assemble in seeds by Ostwald ripening and finally the crystalline core growth occur with remaining solvated ions. Moreover, Ji et al. has proposed a pH-dependant model to streamline the nanospheres final diameter according to UV-visible records and TEM images of intermediates states.13 Two different reaction pathways were identified. The first one that occurred for the acidic pH range in a three steps process: fast nucleation, random attachment to nanowires, and intra-particle ripening. The second pathway under high pH is in line with the nucleation-growth model.
Turkevich–Frens synthesis due to its unchallenged control over size and shape remains a method of choice for the obtaining of water soluble GNPs. Nevertheless citrates that act as reducing agent and passivation ligands are commonly exchanged after the synthesis with thiols in order to enhance the stabilization.14 Several molecules have been tested to replace citrate as reducing agent and stabilizer: carboxylic acids,15 amines,16 polysaccharides,17 thiophene derivatives18 and polymers.19,20 Recently, more exotic synthesis have also been described using peptides21 or plant extracts derivatives22–25 mainly based on catechol functionalities.26–28 In this quest for molecules that both reduce Au(III) ions and act as stabilizers to protect particles against aggregation but also in order to control their functional properties, we were interested in molecules of the 1-hydroxy-1,1-methylene bisphosphonic family (HMBP). These compounds have been used as biocompatible, negatively charged stabilizing agents on superparamagnetic iron oxide (SPIO) nanoparticles29,30 since several years. Moreover functionalized HMBPs have shown to be of great interest for the carbodiimide or click coupling of various compounds on NPs surface.31–35 One of the molecules of this family called Alendronate bearing an amine functionality has also already been used to stabilize (via amine function) GNPs after ligand exchange.36–39 It was even used to synthesize GNPs but the mechanism of action was not elucidated.40 Here we wanted to study more precisely the suitability of such compounds for the synthesis of GNPs in water in a similar manner to the citrate methodology. We expected that HMBP bearing functionality could reduce Au(III) ions and act as an efficient stabilizer of the formed GNP in water and last but not least allow direct functionalization of the surface. Hence, we explored the use of HMBPene as a bifunctional chelating agent for a one-pot synthesis of water soluble GNPs. We precisely studied the mechanism of GNPs formation and the parameter influencing size control. Moreover, in order to have a precise control over crucial physical parameters such as temperature, reaction time and stirring rate,41 we used microwave irradiation to perform the GNPs synthesis.
Experimental part
Materials and apparatus
All water was distilled and subsequently purified to Millipore Milli-Q quality. All glassware used was cleaned with concentrated nitric acid (60%), then rinsed thoroughly with water before use.UV-Visible Absorption spectroscopy of GNPs dipersions was recorded on a Jasco V630 spectrophotometer at neutral pH.
All experiments under microwave (MW) irradiations were run in a 30 mL MW tube, using an Anton Paar monowave 300 microwave synthesis reactor.
1H NMR spectra (400 MHz), proton-decoupled 13C NMR spectra (100.6 MHz) and proton decoupled 31P spectra (162.0 MHz) were recorded on a Bruker Avance 400 spectrometer and chemical shifts are reported in parts per million (ppm) on the δ scale.
High Resolution Mass Spectrometry (HR-MS) experiments were realized on a LTQ Orbitrap Velos (Thermo Scientific) in negative mode using an ESI source. MS spectra were recorded in the Orbitrap mass analyzer allowing a mass accuracy around 1–2 ppm.
DLS and ζ potential analyses were performed on a nano ZS (red badge) ZEN3600 Zetasizer at neutral pH.
TEM images were taken on a FEI CM10 electron microscope. Samples were prepared by dropping of the colloidal solution onto holey carbon-coated Cu grid 10 times.
FTIR absorption spectra were recorded on a Nicolet 380 Thermo Scientific FTIR spectrophotometer. Concentrated GNPs colloidal solution (neutral pH, pre-washed by ultrafiltration) was added to analytical grade KBr and left dried at 80° overnight. The pellet was prepared with this crude powder.
The Thermo Gravimetric Analysis (TGA) curves were recorded using a LabsSys evo TG-DTA-DSC 16000 device manufactured by Setaram Instrumentation.
X-ray Photoelectron Spectroscopy (XPS) data were recorded on a PHI Versaprobe 5000 device and Al–K monochromated radiation (1486.6 eV, 50 W with a 200 μm diameter spot size) was used as X-ray source. Pass energy is 200 eV for spectra and 60 eV for windows (quantifications and curve fitting are obtained from windows acquisitions). Powder samples were prepared by pressing the sample powder on an indium sheet. Neutralization was used to minimize charge effects and the adventitious carbon C1s peak at 284.5 eV was used as the reference. The pressure in analysis chamber during acquisition is around 8.10–8 Pa.
HMBPene synthesis
593 mg (5 mmol) of 4-pentenoyl chloride (ClCO(CH2)2CHCH2, 98%, Sigma-Aldrich, St Louis, MO) were frozen in a round bottom flask of 25 mL with liquid nitrogen, then 5 mL of tris(trimethylsilyl phosphite) (P(OSiMe3)3, 92%, Acros Organics) were added. A white slurry was obtained and stirred over night. Unreacted material was evaporated under vacuum at 70 °C (0.1 Torr) for 20 minutes and hydrolyzed 4 h in 20 mL of MeOH. The solvent was removed under reduced pressure, and the remaining yellow oil was crystallized at pH 2.3 in a MeOH–H2O 9:1 system. The white solid was filtered on Buchner yielding (70%) 940 mg (3.5 mmol) of HMBPene.
1H NMR: δ = 5.90 (m, 1H, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2); 5.02 (dd, Jtrans (H–H) = 17.2 Hz, Jcis (H–H) = 10.2 Hz, 2H, CCH2); 2.32 (t, 3H, CH2COH); 1.99 (dt, 3H, CH2CH2COH).
CH2); 5.02 (dd, Jtrans (H–H) = 17.2 Hz, Jcis (H–H) = 10.2 Hz, 2H, CCH2); 2.32 (t, 3H, CH2COH); 1.99 (dt, 3H, CH2CH2COH).
13C NMR (100.63 MHz, 25 °C, D2O): δ = 139.5; 114.3; 73.8 (t, J1(P–C) = 135.5 Hz, COH); 32.8; 28.0.
HR-MS (ESI-Q Tof) C5H11O7P2: m/z (M − H)−: 244.99818; calc.: 244.998.
31P NMR (161.98 MHz, 25 °C, D2O): δ = 18.9 (s).
IR (KBr, pH7): 2920, 2852, 1640, 1448, 1163, 1107, 907.
General procedure for Au@HMBPene synthesis ([Au] = 0.25 mM, HMBPene–HAuCl4 = 4.4)
Two precursor solutions were prepared: solution A-38.8 mg of chloroauric acid (HAuCl4·3H2O, ≥99.9%, Sigma-Aldrich, St Louis, MO) were dissolved in 5 mL of deionized (DI) water. Solution B-53.6 mg of HMBPene were dissolved in 4.5 mL of DI water, then adjusted to pH 8.2. 125 μL of solution A and 250 μL of solution B were added to 9.5 mL of DI water in a 30 mL MW tube. The mixture was heat at 100 °C during 10 min. A red wine coloured solution of 14.4 ± 1.0 nm diameter GNPs was obtained.General procedure for Au@Ct synthesis ([Au] = 0.25 mM, CtNa3–HAuCl4 = 4.4)
Two precursor solutions were prepared: solution A-38.8 mg of chloroauric acid (HAuCl4·3H2O, ≥99.9%, Sigma-Aldrich, St Louis, MO) were dissolved in 5 mL of DI water. Solutions B-59.1 mg of trisodium citrate (Na3C6H5O7, ≥99%, Acros Organics) were dissolved in 5 mL of DI water. 125 μL of solution A and 250 μL of solution B were added to 9.5 mL of DI water in a 30 mL MW tube. The mixture was heat at 100 °C during 10 min. A red wine coloured solution of 14.1 ± 1.1 nm diameter GNPs was obtained. 25 μL of solution B were added to this Au@Ct NPs solution in order to adjust the citrate concentration on previously described HMBPene–HAuCl4 ratio.Stability test against cysteamine hydrochloride (Au@HMBPene vs. Au@Ct)
100 μL of Cysteamine hydrochloride (HSCH2CH2NH2·HCl, ≥98%, Sigma-Aldrich, St Louis, MO) solution (0 μM, 10 μM, 20 μM and 40 μM) were added to 2 mL of 14 nm GNPs solution (freshly synthesized following the general procedure). UV spectra were registered after 10 min.Results and discussion
Microwave assisted synthesis of Au@HMBPene nanoparticles in water
The HMBPene was synthesized according to a methodology already described31 from condensation of pentenoyl chloride with tris(trimethylsilyl) phosphite. This compound was fully characterized by NMR (see ESI, Fig. S1–S2†) and mass spectrometry. This work proposes to use the HMBPene as the reductant and stabilizer for GNPs synthesis within one step process. In order to control precisely the heating of the solution and get reproducible synthesis we evaluated the GNPs synthesis under MW conditions. First of all the Turkevich–Frens classical synthesis was adapted to MW conditions by mixing at room temperature HAuCl4 and citrate and heating 10 min. at 100 °C. Concerning the spherical shape and the size distribution of the obtained nanoparticles, no major differences between classical conditions and microwave conditions were observed (see ESI, Fig. S5†). Therefore the MW method appears to be a more convenient method compared to the classical one because the two reagents were mixed together at room temperature before heating. We performed the first experiment using HMBPene instead of citrate with similar conditions compared to Ji et al. work. Hence, HAuCl4 concentration was fixed to 0.25 mM, a HMBPene–HAuCl4 ratio at 4.4 was used and the initial pH of HMBPene solution fixed at 8.2. After 10 min microwave heating at 100 °C, the resulting wine-red solution obtained has similar characteristics to the citrated GNPs synthesized (prepared with classical conditions or MW conditions) (Fig. 1). The formed gold nanoparticles presented a relatively narrow size distribution with a mean diameter of 14.4 ± 1.0 nm, as depicted on Fig. 1, proving the homogeneity of the nucleation process. The Au@HMBPene nanoparticles formed highly stable water dispersion. At neutral pH, the Z-average diameter and zeta potential surface are found equal to 16.5 nm and −38 mV respectively. Considering mean crystalline core (14 nm in diameter) and a layer of HMBPene, this suggested very low aggregation of the synthesized materials. The colloidal stability is mainly due to electrostatic repulsion between the negatively charged GNPs, induced by the HMBP moieties. No aggregates were observed by DLS over one month (see ESI, Fig. S6†). The SPR band in the UV visible region has a maximum absorption at λ = 516 nm, which is in good accordance for this scale range, according to Haiss et al.42 This particles synthesis was scaled up until a gold concentration of 1 mM with minor changes of obtained physical properties (see ESI, Fig. S7†). | ||
| Fig. 1 Au@HMBPene NPs (a) synthesis, (b) TEM image, (c) TEM size distribution (bar chart), Z-average diameter (blue bar) and (d) UV-visible absorption. | ||
In order to understand the process by which HMBPene allow the formation of GNPs the colloid solution just after reaction was analyzed by NMR (Fig. 2). On the 31P NMR spectrum the signal corresponding to the HMBPene remains the most intensive one but the apparition of several peaks belonging to PO4R3 species could be observed in the −10 to 5 ppm region and many shifted peaks around 18 ppm corresponding to phosphonate groups with an electronically modified environment were discernable. So it can be reasonably hypothesized that the chemical reduction of Au(III) complex occurred mainly because of the oxidation of the phosphonate group to phosphate group. This can happened by degradation of the P–C–P bridge. Though extremely stable in bisphosphonic acid form such degradation was already observed for bisphosphonate esters in basic conditions through phosphono-phosphate isomerisation.43 Moreover it should be noted that such formation of phosphoric adducts is one of the fragmentation pathways that can be observed when studying by mass spectrometry several bisphosphonic structures.44–46 As the 13C and 1H NMR did not show significant modifications compared to the starting compound (see ESI, Fig. S3–S4†), we can conclude that the carbon skeleton has not reacted.
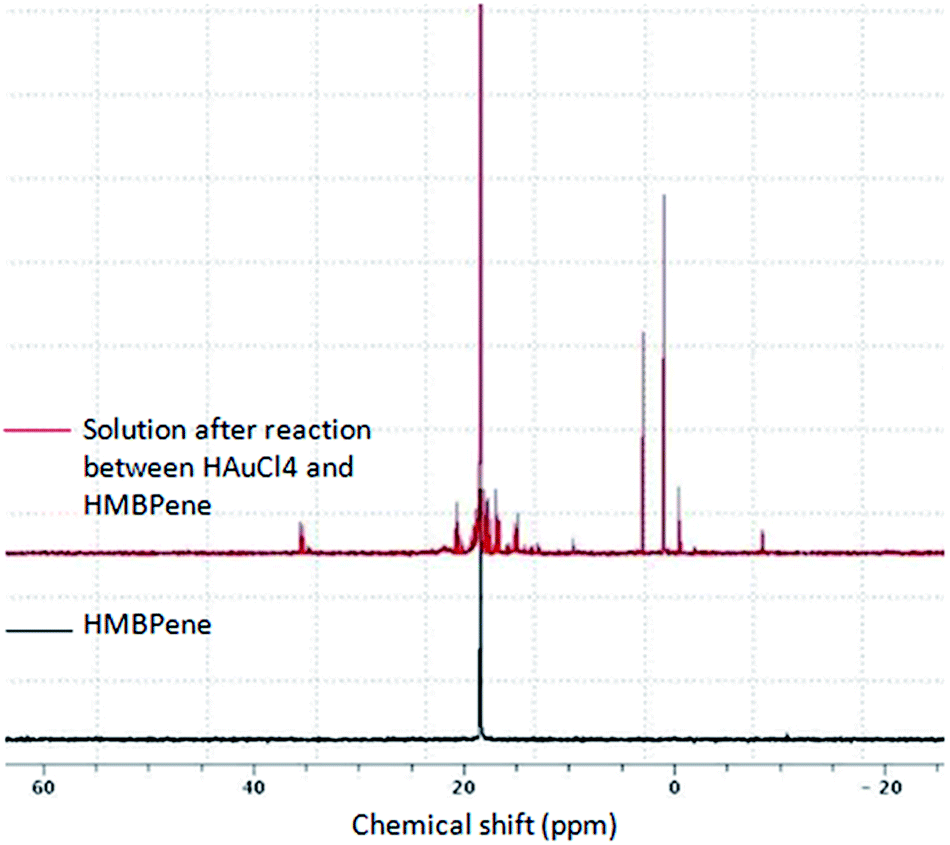 | ||
| Fig. 2 31P NMR (300 MHz) spectra of HMBPene partial degradation. | ||
Surface characterization
HMBPene and by-products can be removed by ultrafiltration, then GNPs are dispersed in pure water and analyzed by FTIR spectrometry (Fig. 3). The chemisorption of HMBPene was qualitatively assessed comparing the coated nanocrystals (purple curve) with the free HMBPene molecule (red curve). Large modifications are observed within the PO region (900–1200 cm−1). The strong tightening of PO and P–O vibration bands around 1000 cm−1 (purple curve) which is characteristic of the chelation of phosphorus species on a metallic surface,29,34 suggested a coordination of phosphonates as chelating groups. Contrary to the previous work of Drogat et al.,40 it appears that the bisphosphonate function is coordinated to the gold surface. The observed modifications in the PO region also suggested that no free phosphonate functions were present at the surface, ruling out the possible formation of a bilayer of HMBP with ethylenic part in the middle. Further evidence of a monolayer formation was given by TGA (see ESI, Fig. S8†) that indicated a surface coverage of 2.2 HMBPene per nm2 which is in complete accordance to the results already observed for formation of HMBP monolayer onto iron oxide nanoparticles.32 Vibration bands of C–H stretching (2920–2852 cm−1), C–H scissoring (1448 cm−1) and CC stretching (1640 cm−1) remained unmodified compared to free HMBPene. We pointed out that C–H stretching signals were enhanced when HMBPene was adsorbed on GNPs surface, but also CH2 deformation (1391 cm−1) instead of C–H scissoring. These results are in accordance with the observation of the phosphonate binding on the gold surface and the double bond CC remained unmodified after synthesis. Hence ethylenic functions are still available as a reactive group on the nanoplatform.
 | ||
| Fig. 3 FTIR analysis: HMBPene (red spectrum) vs. GNPs surface (purple spectrum). | ||
XPS analysis of the isolated HMBPene showed that binding energy of P2p electron is about 132.8–133.9 eV (Fig. 4). GNPs powder has a P2p peak around 132.5–133.6 eV. Such a difference could be related to the fact that oxygen withdrawing effect on the phosphorus atom is declined when phosphonic functions are engaged in coordination bonding to the metal. Basly et al. has observed the same effect for phosphonate binding to iron oxide surface.47 Moreover the P2p peak width of Au@HMBPene is lower than the HMBPene one, proving that phosphorus electronic environment has changed. Therefore we propose a phosphonate chelation of the NP surface according to FTIR data and XPS data.
 | ||
| Fig. 4 Binding energy of phosphonate P2p electrons: Au@HMBPene (a) vs. HMBPene (b). | ||
pH effect on final size of Au@HMBPene NPs: analogy with the Turkevitch–Frens synthesis
As already mentioned, in Turkevich GNPs synthesis pH effect is described to have crucial importance on the GNPs growth mechanism and therefore on the final size or shape of the nanoparticles. So, we studied the pH solution (Fig. 5). Firstly the initial gold chloride concentration of the solution was unchanged and the concentration of HMBPene at constant pH was varied (Fig. 5a and see ESI, Fig. S9†). Secondly we kept both gold chloride and HMBPene concentration unchanged and varied the pH of HMBPene solution (Fig. 5b). Fig. 5 also presents the results of these two experiments with final pH in ordinate rather the final diameter (Fig. 5c and d). The first study (Fig. 5a–c) clearly showed the same trend as the Turkevich–Frens synthesis (Fig. 5a and c captions). Therefore Au@HMBPene NPs were formed through the two pathways described by Ji et al. for citrate capped GNPs: (i) fast nucleation – random attachment-ripening (ii) nucleation-growth. On the other hand when studying the effect of pH of HMBPene solution on final diameter (Fig. 5b or final pH Fig. 5d) the obtained results could also be related to the first study. Hence These results were comparable to the effect of HMBPene–HAuCl4 ratio from 2:1 to 4.4:1 (inserted graphs of Fig. 5b and d). When varying pH of the precursor solution from 4.2 to 11.8 one must note that obtained GNPs perfectly kept their spherical aspect with narrow size distributions (Fig. 6).
 | ||
| Fig. 5 Summary of the average sizes of GNPs using (a) different HMBPene–HAuCl4 precursor ratios compared to Ji et al. Work (captions, adapted with permission from (X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939–13948). Copyright (2007) American Chemical Society) and (b) different initial HMBPene solution pH compared to enlarged graph (a) corresponding to dotted square. Summary of final pH were reported using (c) different HMBPene–HAuCl4 precursor ratios compared to Ji et al. Work (captions, adapted with permission from (X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939–13948). Copyright (2007) American Chemical Society) and (d) different initial HMBPene solution pH compared to enlarged graph (c) corresponding to dotted square. All of these experiments have been done with the general method described in the experimental part (solid lines are a guide to the eye). | ||
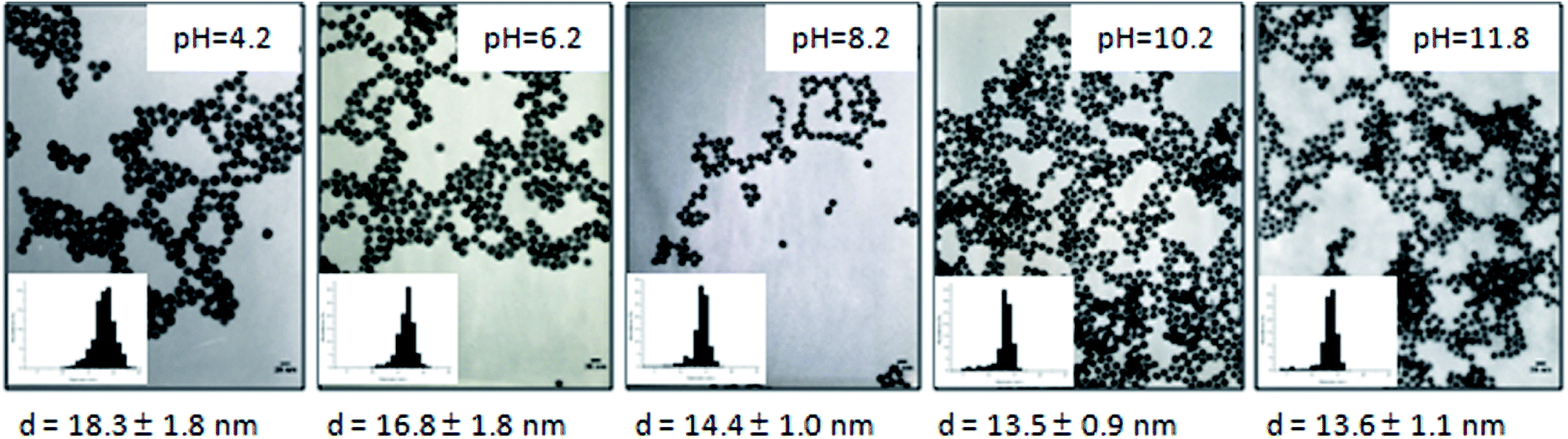 | ||
| Fig. 6 TEM images of GNPs synthesized by varying the HMBPene solution pH and corresponding size distribution. All of these experiments have been done with the general method described in the experimental part. | ||
When decreasing the HMBPene–HAuCl4 ratio below 2, obtained GNPs gradually lost their sphericity. For a ratio HMBPene–HAuCl4 = 1, slightly elongated GNPs (“nanorice”) were obtained and further decrease of the ratio (0.9 and 0.8) were correlated to an increase of the phenomenon together with an increase of size and polydispersity (see ESI, Fig. S10†). This behavior could be correlated to a default in ligands amount during the crystal growth. Interestingly, nanowires were obtained for HMBPene–HAuCl4 = 0.7 as showed by TEM images (Fig. 7a and b). The UV-visible spectra (Fig. 7c) showed a broad peak in the 500–700 nm range with low absorption intensity. This spectral signature is a feature of the gold nanowires anisotropic nature as observed by Ji et al. for Ct–HAuCl4 = 0.7 until 300 seconds of reaction. This result suggests that the intra-particle ripening step was unfinished after 10 min at 100 °C for this ratio. This fact is once again in perfect agreement with the proposed nucleation–aggregation-smoothing model for acidic conditions.13
 | ||
| Fig. 7 TEM images (a and b) and UV-visible absorption (c) for unachieved reaction for a precursor ratio of (0.7:2). | ||
Stability tests against cysteamine hydrochloride–HMBPene vs. citrate
Cysteamine as water soluble thiolate molecule was used to differentiate citrate and HMBPene GNPs by their binding strength to the gold surface. It is widely known that thiolate compounds are able to link gold more strongly than oxygen chelating groups48 according to the HSAB (Hard and Soft Acids and Bases) theory. Therefore, a ligand exchange proceeds by introducing a thiol solution in aqueous GNPs dispersions stabilized with carboxylate groups. Cysteamine hydrochloride brings a positive charge on the NP surface and decreasing the absolute negative charges. This reduces electrostatic repulsion between GNPs and leading to aggregation. Varied load of Cysteamine hydrochloride were added to several samples of previously described GNPs dispersions and spectrum were recorded after 10 minutes of stirring (no more colour changes were seen over this time Fig. 8).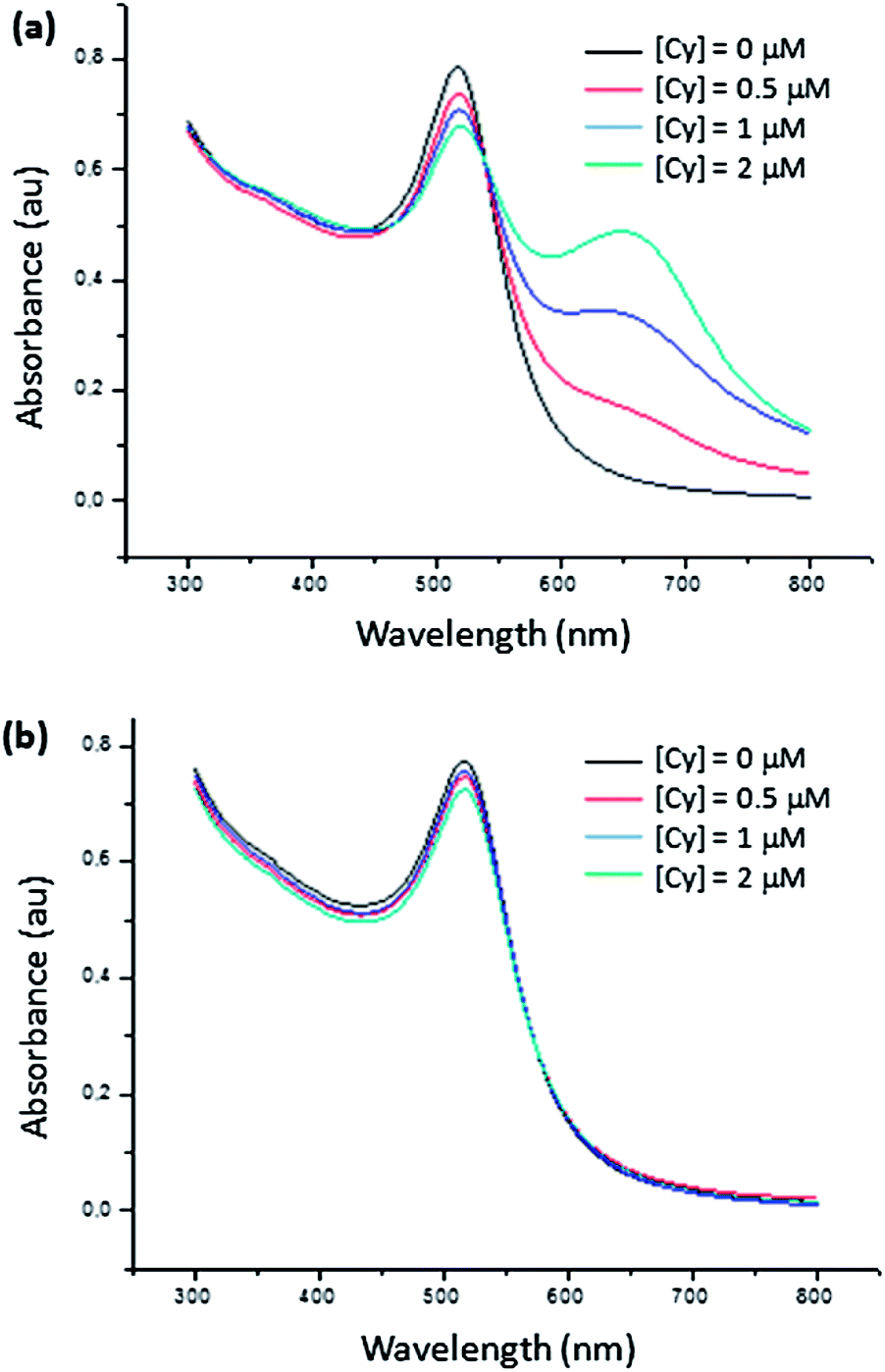 | ||
| Fig. 8 Absorption spectra of (a) Au@Ct and (b) Au@HMBPene after adding cysteamine hydrochloride (Cy) at different concentrations. | ||
Unambiguously, Au@HMBPene NPs remained stable whereas Au@Ct NPs were partially aggregated. This means that the thermodynamic aspect of exchange reactions is correlated to the strength bonding interaction difference between phosphonate group and carboxylate group. Therefore HMBPene appears to be a more efficient stabilizer than citrate against ligand exchange. This advantage of Au@HMBPene NPs constitutes a key property for further functionalization on the terminal alkene group.
Conclusions
A new GNPs synthesis was performed in one-pot protocol including MW assisted gold reduction, coating and functionalization steps using HMBPene. GNPs were well-dispersed, stable and isolated in water. Changing the reaction pH allowed an accurate control of GNPs size following the Turkevich–Frens synthesis mechanism proposed by X. Ji et al. The GNPs shape remains spherical on a large pH scale variation. These nanocrystals surface present an ethylenic bond, which can be a reactive site for further functionalization. Hence, bisphosphonate chemistry appears to be a promising tool for the development of functionalized gold nanoplatforms.Acknowledgements
Our special thanks to Julie Hardouin for her excellent technical assistance in mass spectroscopy.Notes and references
- M.-C. Daniel and D. Astruc, Chem. Rev., 2003, 104, 293–346 CrossRef PubMed.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS PubMed.
- M. R. Jones, K. D. Osberg, R. J. Macfarlane, M. R. Langille and C. A. Mirkin, Chem. Rev., 2011, 111, 3736–3827 CrossRef CAS PubMed.
- A. J. Mieszawska, W. J. M. Mulder, Z. A. Fayad and D. P. Cormode, Mol. Pharm., 2013, 10, 831–847 CrossRef CAS PubMed.
- C. J. Murphy, T. K. Sau, A. M. Gole, C. J. Orendorff, J. Gao, L. Gou, S. E. Hunyadi and T. Li, J. Phys. Chem. B, 2005, 109, 13857–13870 CrossRef CAS PubMed.
- B. Kang, L. A. Austin and M. A. El-Sayed, ACS Nano, 2014, 8, 4883–4892 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- G. Frens, Nature, 1973, 241, 20–22 CAS.
- S. Kumar, K. S. Gandhi and R. Kumar, Ind. Eng. Chem. Res., 2006, 46, 3128–3136 CrossRef.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- B.-K. Pong, H. I. Elim, J.-X. Chong, W. Ji, B. L. Trout and J.-Y. Lee, J. Phys. Chem. C, 2007, 111, 6281–6287 CAS.
- J. Polte, T. T. Ahner, F. Delissen, S. Sokolov, F. Emmerling, A. F. Thünemann and R. Kraehnert, J. Am. Chem. Soc., 2010, 132, 1296–1301 CrossRef CAS PubMed.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939–13948 CrossRef CAS PubMed.
- K. Siriwardana, M. Gadogbe, S. M. Ansar, E. S. Vasquez, W. E. Collier, S. Zou, K. B. Walters and D. Zhang, J. Phys. Chem. C, 2014, 118, 11111–11119 CAS.
- S. A. Moreno-Álvarez, G. A. Martínez-Castañón, N. Niño-Martínez, J. F. Reyes-Macías, N. Patiño-Marín, J. P. Loyola-Rodríguez and F. Ruiz, J. Nanopart. Res., 2010, 12, 2741–2746 CrossRef.
- H. Zhu, Z. Pan, E. W. Hagaman, C. Liang, S. H. Overbury and S. Dai, J. Colloid Interface Sci., 2005, 287, 360–365 CrossRef CAS PubMed.
- C. Fan, W. Li, S. Zhao, J. Chen and X. Li, Mater. Lett., 2008, 62, 3518–3520 CrossRef CAS PubMed.
- E. Ventosa, A. Colina, A. Heras, V. Ruiz, J. Garoz and J. López-Palacios, J. Nanopart. Res., 2012, 14, 1–10 CrossRef.
- P. Abdulkin, T. L. Precht, B. R. Knappett, H. E. Skelton, D. A. Jefferson and A. E. H. Wheatley, Part. Part. Syst. Charact., 2014, 31, 571–579 CrossRef CAS.
- E. B. Ferreira, J. F. Gomes, G. Tremiliosi-Filho and L. H. S. Gasparotto, Mater. Res. Bull., 2014, 55, 131–136 CrossRef CAS PubMed.
- Y. Li, Z. Tang, P. N. Prasad, M. R. Knecht and M. T. Swihart, Nanoscale, 2014, 6, 3165–3172 RSC.
- S. S. Shankar, A. Ahmad, R. Pasricha and M. Sastry, J. Mater. Chem., 2003, 13, 1822–1826 RSC.
- X. Jiang, D. Sun, G. Zhang, N. He, H. Liu, J. Huang, T. Odoom-Wubah and Q. Li, J. Nanopart. Res., 2013, 15, 1–11 CrossRef.
- S. Sivaraman, S. Kumar and V. Santhanam, Gold Bull., 2010, 43, 275–286 CrossRef CAS.
- M. N. Nadagouda, N. Iyanna, J. Lalley, C. Han, D. D. Dionysiou and R. S. Varma, ACS Sustainable Chem. Eng., 2014, 2, 1717–1723 CrossRef CAS.
- S. C. Sahu, A. K. Samantara, A. Ghosh and B. K. Jena, Chem.–Eur. J., 2013, 19, 8220–8226 CrossRef CAS PubMed.
- K. C. L. Black, Z. Liu and P. B. Messersmith, Chem. Mater., 2011, 23, 1130–1135 CrossRef CAS PubMed.
- S. Aswathy Aromal and D. Philip, Phys. E, 2012, 44, 1692–1696 CrossRef CAS PubMed.
- Y. Lalatonne, C. Paris, J. M. Serfaty, P. Weinmann, M. Lecouvey and L. Motte, Chem. Commun., 2008, 2553–2555 RSC.
- L. Motte, F. Benyettou, C. de Beaucorps, M. Lecouvey, I. Milesovic and Y. Lalatonne, Faraday Discuss., 2011, 149, 211–225 RSC.
- P. Demay-Drouhard, E. Nehlig, J. Hardouin, L. Motte and E. Guénin, Chem.–Eur. J., 2013, 19, 8388–8392 CrossRef CAS PubMed.
- J. Bolley, E. Guenin, N. Lievre, M. Lecouvey, M. Soussan, Y. Lalatonne and L. Motte, Langmuir, 2013, 29, 14639–14647 CrossRef CAS PubMed.
- E. Nehlig, L. Motte and E. Guénin, Catal. Today, 2013, 208, 90–96 CrossRef CAS PubMed.
- F. Benyettou, E. Guénin, Y. Lalatonne and L. Motte, Nanotechnology, 2011, 22, 055102 CrossRef CAS PubMed.
- E. Guénin, J. Hardouin, Y. Lalatonne and L. Motte, J. Nanopart. Res., 2012, 14, 1–10 CrossRef.
- R. Ross, L. Cole and R. Roeder, J. Nanopart. Res., 2012, 14, 1–11 CrossRef.
- R. D. Ross, L. E. Cole, J. M. R. Tilley and R. K. Roeder, Chem. Mater., 2014, 26, 1187–1194 CrossRef CAS.
- R. D. Ross and R. K. Roeder, J. Biomed. Mater. Res., Part A, 2011, 99, 58–66 CrossRef PubMed.
- L. E. Cole, T. Vargo-Gogola and R. K. Roeder, Biomaterials, 2014, 35, 2312–2321 CrossRef CAS PubMed.
- N. Drogat, L. c. Jauberty, V. Chaleix, R. Granet, E. Guénin, V. Sol and V. Gloaguen, Mater. Lett., 2014, 122, 208–211 CrossRef CAS PubMed.
- N. Dahal, S. Garcìa, J. Zhou and S. M. Humphrey, ACS Nano, 2012, 6, 9433–9446 CrossRef CAS PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- P. A. Turhanen and J. J. Vepsäläinen, Beilstein J. Org. Chem., 2008, 4, 7 CrossRef PubMed.
- J. Hardouin, E. Guénin, C. Malosse, M. Caron and M. Lecouvey, Rapid Commun. Mass Spectrom., 2008, 22, 2287–2300 CrossRef CAS PubMed.
- J. Hardouin, E. Guénin, M. Monteil, M. Caron and M. Lecouvey, J. Mass Spectrom., 2008, 43, 1037–1044 CrossRef CAS PubMed.
- E. Guénin, M. Lecouvey and J. Hardouin, Rapid Commun. Mass Spectrom., 2009, 23, 1234–1240 CrossRef PubMed.
- B. Basly, G. Popa, S. Fleutot, B. P. Pichon, A. Garofalo, C. Ghobril, C. Billotey, A. Berniard, P. Bonazza, H. Martinez, D. Felder-Flesch and S. Begin-Colin, Dalton Trans., 2013, 2146–2157 RSC.
- R. G. Acres, V. Feyer, N. Tsud, E. Carlino and K. C. Prince, J. Phys. Chem. C, 2014, 118, 10481–10487 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional HMBPene and GNPs characterizations. See DOI: 10.1039/c4ra11847b |
| This journal is © The Royal Society of Chemistry 2014 |