
Dual application of Pd nanoparticles supported on mesoporous silica SBA-15 and MSU-2: supported catalysts for C–C coupling reactions and cytotoxic agents against human cancer cell lines†
Adriana Balbína,
Francesco Gaballoa,
Jesús Ceballos-Torresa,
Sanjiv Prashara,
Mariano Fajardoa,
Goran N. Kaluđerović*b and
Santiago Gómez-Ruiz*a
aDepartamento de Biología y Geología, Física y Química Inorgánica, Escuela Superior de Ciencias Experimentales y Tecnología, Universidad Rey Juan Carlos, Calle Tulipán s/n, E-28933, Móstoles, Madrid, Spain. E-mail: santiago.gomez@urjc.es
bDepartment of Bioorganic Chemistry, Leibniz Institute of Plant Biochemistry, Weinberg 3, D-06120 Halle (Saale), Germany. E-mail: goran.kaluderovic@ipb-halle.de
First published on 17th October 2014
Abstract
Two different mesoporous silica-based materials (SBA-15 and MSU-2) have been treated under mild conditions with different quantities of [PdCl2(cod)] (cod = 1,5-cyclooctadiene) to promote the formation of supported palladium nanoparticles (materials of the type SBA-15–Pd and MSU-2–Pd). The synthesized materials have been characterized by different techniques observing that the palladium nanoparticles remain impregnated in the silica. The catalytic activity of the hybrid Pd–silica materials has been tested in Suzuki–Miyaura C–C coupling reactions observing moderate conversion rates in the reactions of 3-bromoanisole with 4-carboxyphenylboronic acid and 2-bromopyridine with 4-carboxyphenylboronic acid. In addition, the synthesized materials showed a good degree of recyclability, being catalytically active in five consecutive catalytic tests. Finally, in order to evaluate the cytotoxicity of the synthesized materials, in vitro tests against five different human cancer cell lines have been carried out, observing high cytotoxic activities of the hybrid systems comparable if not somewhat higher to other systems based on metal complexes supported on mesoporous silicas described previously in the literature. To the best of our knowledge the cytotoxic study reported here represents the first evaluation of the anticancer action of supported palladium nanoparticles in human cancer cells.
1. Introduction
Metal nanoparticles have attracted much attention over the last decade in a variety of applications including sensors, non-linear optics, medical dressings and catalysis.1–4 The supporting material on which these nanoparticles are synthesized has also received significant scrutiny in the literature.1 The choice of solid supports in the synthesis of impregnated metal nanoparticles is very wide, ranging from organic polymers5 to various transition metal oxides,6 and including mesoporous silicas1 and microporous zeolites.7 However, the most important factors to consider when choosing the support material are its thermal and chemical stability under the reaction conditions and its ability for facile dispersion and the accessibility of the metal nanoparticles (this is generally achieved with materials with high surface areas (>100 m2 g−1) and mesoporous structures (pore size >2 nm)).1 In this context, mesoporous silicas have been considered to be very good supports for metal nanoparticles. Mesoporous silicas were discovered in the early 1990s by scientists at Mobil Oil Company and by Kuroda and co-workers, in part as a response for the need of extending the applications of zeolites.8 These interesting materials offer many of the features expected from an ideal support: a very large surface area (>600 m2 g−1), a narrow pore distribution (2–50 nm), high thermal and chemical stability, ready availability and easy chemical modification on the surface by anchoring catalytically active species. Due to their properties, mesoporous silicas are actively being exploited as supports in catalysis. The presence of functional groups and active species on the surface (thiol groups, organic ligands, metal nanoparticles, etc.) has a great influence on the recyclability of the catalyst, on its activity and on the agglomeration of the nanoparticles.1The physical properties and catalytic activity of supported catalysts are highly dependent on the support, methods of preparation and type and method of preparation of the noble metal particles.9 The incorporation and position of palladium nanoparticles in the final hybrid material and the maximum effective loading of metal nanoparticles are aspects that must be considered in the nanoparticle preparation. Finally, the control of size and morphology of the nanoparticles and how to maintain it during the catalytic reaction are two main problems that remain unsolved. In addition, the presence of certain functional groups on the surface of the supports may have an important role regarding these aspects, as well as others such as catalytic activity or controlling the phenomenon of particle agglomeration.9
Currently, supported catalysts are prepared in two ways: directly during surface synthesis or by post-synthesis treatment of the surface with a palladium precursor. The post-synthetic methods are normally based on impregnation of a Pd precursor (e.g. palladium acetate) followed by its reduction with various reducing agents (ethanol, NaBH4, H2, hydrazine),1,2,9 ion exchange with metal precursors,10 adsorption of molecular cluster precursors,11 immobilization of metal complexes12 or photocatalytic reduction.13 The method used for the synthesis of the nanoparticles on the support, inside the pores as well as on its external surface, affects their distribution and size in a decisive manner. For example, it has been found that if the synthesis of Pd nanoparticles occurs simultaneously to the synthesis of the silica support MCM-41, the nanoparticles remain anchored to the outer surface, while if they are introduced through a post-synthesis treatment they can be incorporated within the mesoporous structure of MCM-41.14 In addition, several reports have been published using SBA-15 mesoporous materials as scaffolds for the preparation of palladium nanoparticles15–17 with similar results to those described for MCM-41, however, nothing is known about the use of other mesoporous silica with disordered pores such as MSU-2, in the preparation of such hybrid systems.
All the cited methods are fast and simple, but when the catalyst is reused, the activity decreases dramatically due to its structure collapsing.18 Additionally, the kinetics of the metal reduction process, in most cases, is not easily controllable, nor green nor simple to carry out; the product also needs to be thoroughly washed and conditioned in order to remove the excess of reducing agent. This has led to a high number of reports concerning improvements in the preparation methods of metal nanoparticles, and recently, some research groups have employed glucose as an environmentally benign reducing agent to prepare well-dispersed metal nanoparticles with an excellent catalytic activity in C–C cross coupling reactions and a very easy recyclability.2 Additionally, many other groups have used a previous functionalization of the mesoporous silica with amino or thiol groups for an easier reduction of the metal salt and in situ formation of the metal nanoparticle.19–22
Thus, metal nanoparticles supported on mesoporous silicas have shown interesting catalytic properties, however, at the present time only very limited reports have analyzed the cytotoxicity of supported palladium nanoparticles or free palladium nanoparticles.23–26 From other cytotoxic studies with metal oxide nanoparticles one can envisage that nanoparticles influence the cytotoxic effects according to their particle size, surface area and the type and concentration of metal ions released into the medium, being the latter the most important effect on the cytotoxicity.27 Furthermore, it is known that the cytotoxic nature is greater for soluble nanoparticles.27 The development of nanoparticles-based anticancer drugs has become important in the world of biomedicine, since they act only in cancer cells and have fewer side effects. The mechanisms by which these nanoparticles exhibit anticancer activity are not fully understood.27 However, it is known that the key to understanding the nanoparticle cytotoxicity is their small size, lower than that of a cell, which allows them to penetrate the biological structures, altering their normal function. In addition, the nanoparticles side effects on human health, beyond the chemistry, size, shape, charge, agglomeration state and electromagnetic properties, depend on individual factors such as genetic and existing disease,27 although further studies in this topic must be carried out in the future.
Thus, bearing in mind the interest in the catalytic and biological properties of metal nanoparticles, in this paper we report the synthesis, characterization and catalytic properties of palladium nanoparticles supported on SBA-15 and MSU-2 which represents the first study of the preparation of palladium nanoparticles using a mesoporous silica with a non-ordered pore distribution (MSU-2). In addition, the synthetic method described here for the preparation of the hybrid systems does not need a previous functionalization of the mesoporous silica with amino or thiol groups, but a simple reduction of an organometallic salt of Pd such as [PdCl2(cod)] in a single step. The synthesized materials showed moderate conversions and a good degree of recyclability in C–C coupling Suzuki–Miyaura reactions.
In addition, we herein report the preliminary evaluation of the in vitro cytotoxic activity of the hybrid materials against four different human cancer cell lines which shows a dose-dependent activity with M50 values between 110 ± 20 and 553 ± 13 μg mL−1. This represents the first report describing the cytotoxic properties of supported palladium nanoparticles. These studies should be useful as a starting point for subsequent investigations of palladium nanoparticles with biological properties.
2. Experimental
2.1. General conditions
All manipulations were performed under dry nitrogen gas using standard Schlenk techniques and dry box. Solvents were distilled from the appropriate drying agents and degassed before use. Tetraethylorthosilicate (TEOS) 98% (MW = 208.33, d = 0.934 g mL−1), poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (Pluronic 123, Mav = 5800; d = 1.019 g mL−1), NaF (extra pure) and Tergitol® NP-9 (MW = 616.82), all from Sigma-Aldrich, were used as purchased, without further purification. Water (resistance 18.2 MΩ cm) used in the preparation of materials was obtained from a Millipore Milli-Q-System (Billerica, MA, USA).2.2. General remarks on the characterization of the materials
X-ray diffraction (XRD) pattern of the silicas were obtained on a Philips Diffractometer model PW3040/00 X'Pert MPD/MRD at 45 kV and 40 mA, using a wavelength Cu Kα (λ = 1.5418 Å). Pd wt% determination by X-ray fluorescence were carried out with a X-ray fluorescence spectrophotometer Philips MagiX with an X-ray source of 1 kW and a Rh anode using a helium atmosphere. The quantification method is capable of analyzing from 0.0001% to 100% Pd. N2 gas adsorption–desorption isotherms were performed using a Micromeritics ASAP 2020 analyzer. Scanning electron micrographs and morphological analysis were carried out on a XL30 ESEM Philips with an energy dispersive spectrometry system (EDS). The samples were treated with a sputtering method with the following parameters: sputter time 100 s, Sputter current 30 mA film thickness 20 nm using a Sputter coater BAL-TEC SCD 005. Conventional transmission electron microscopy (TEM) was carried out on a TECNAI 20 Philips, operating at 200 kV.2.3. Preparation of non-functionalized materials
Afterwards, the solution was filtered under vacuum and the resulting white solid was abundantly washed with Milli-Q water, in order to eliminate soluble impurities and the remaining surfactant. After washing, a drying process (at 100 °C during 6 h) and a subsequent calcination process (during 24 h at 500 °C) were carried out in a muffle oven. After the calcination process, 29.17 g of fine white powder of SBA-15 was obtained.
2.4. Preparation of the hybrid materials SBA-15–Pd-X or MSU-2–Pd-X
| Material | Starting material (2.0 g) | Theoretical Pd content (%) | Pd (mg) | [PdCl2(cod)] (mg) | Experimental Pd content (%) |
|---|---|---|---|---|---|
| SBA-15–Pd-1 | SBA-15 | 1 | 20 | 54 | 0.39 |
| SBA-15–Pd-2 | SBA-15 | 2 | 40 | 108 | 0.54 |
| SBA-15–Pd-5 | SBA-15 | 5 | 101 | 270 | 1.20 |
| SBA-15–Pd-10 | SBA-15 | 10 | 201 | 540 | 2.10 |
| SBA-15–Pd-20 | SBA-15 | 20 | 388 | 1040 | 6.11 |
| SBA-15–Pd-50 | SBA-15 | 50 | 1006 | 2700 | 12.80 |
| MSU-2–Pd-1 | MSU-2 | 1 | 20 | 54 | 0.63 |
| MSU-2–Pd-2 | MSU-2 | 2 | 40 | 108 | 0.48 |
| MSU-2–Pd-5 | MSU-2 | 5 | 101 | 270 | 0.70 |
| MSU-2–Pd-10 | MSU-2 | 10 | 201 | 540 | 1.12 |
| MSU-2–Pd-15 | MSU-2 | 15 | 294 | 790 | 2.18 |
| MSU-2–Pd-20 | MSU-2 | 20 | 388 | 1040 | 4.46 |
The general procedure carried out for the preparation of the hybrid materials was as follows: in a dry box, the corresponding amount of the mesoporous silica and [PdCl2(cod)] were added to a Schlenk tube and dried under vacuum for 1 h at room temperature. Subsequently, 30 mL of tetrahydrofurane (THF) were added under an inert atmosphere. The reaction mixture was then heated at 80 °C and stirred for 48 h. The reaction conditions were determined after several syntheses at different temperatures, reaction times and solvents, showing that the greater efficacy was obtained using THF as solvent, at 80 °C and reaction time of 48 h. Subsequently, the hybrid material was filtered and washed several times with solvents of increasing volatility (2 × 50 mL each), namely THF, toluene, hexane and diethylether. Finally, the material was dried under vacuum for 12 h to remove all solvent trace. This procedure was repeated for each initial amount of palladium and for all the studied materials (SBA-15 and MSU-2).
2.5. Catalytic tests
(1) Reaction between 3-bromoanisole and 4-carboxyphenylboronic acid (Scheme 1).
 | ||
| Scheme 1 | ||
(2) Reaction between 2-bromopyridine and 4-carboxyphenylboronic acid (Scheme 2).
 | ||
| Scheme 2 | ||
The reactions were performed under identical conditions, in order to facilitate a subsequent analysis of the results. Thus, the limiting reagent was the halide derivative, the molar ratio between the boronic acid and the halide was 1.2 to 0.8, the molar ratio between the base (K2CO3) and the halide was 2 to 1 and the amount of catalyst was always 50 mg. The experimental procedure was identical for both reactions, using the corresponding halide in each case. Degassed solvents and a nitrogen atmosphere were used in the reaction to achieve higher final conversions.22
In a typical catalytic reaction a three-neck flask was filled with boronic acid (0.2 g, 1.2 mmol), K2CO3 (0.22 g, 1.6 mmol) and Pd catalyst and three vacuum/N2 cycles (10 min/1 min) under stirring were applied to remove oxygen from the reaction atmosphere. In parallel, the solvent (DMF–H2O 95/5, 10 mL) and the aryl halide were mixed under nitrogen in a Schlenk tube, bubbling N2 inside the solution for 15 min to eliminate dissolved oxygen. Subsequently, this mixture was transferred under N2 to the three-neck flask with the solid mixture. The suspension was then heated to the corresponding temperature (using a condenser in the case of refluxing conditions) and stirred for 24 h. After this time, the solution was cooled to room temperature and the solvent was removed under vacuum.
The catalytic activity of the synthesized materials in the Suzuki–Miyaura coupling reactions has been compared to the activity of a homogeneous Pd catalyst, [Pd(PPh3)4] using 1% Pd.
2.5.1.1. Purification of the catalytic products. The purification of the product was achieved using a chromatographic column, charged with silica gel and the suitable eluent to separate the reaction product. The latter was chosen on the basis of tests carried out on thin layer chromatography (TLC).
For reaction 1 (3-bromoanisole + 4-carboxyphenylboronic acid), dichloromethane–toluene (9/1) was used as the eluent solution. Solvent was removed from the solution containing the product with a rotavapor and the light yellow solid obtained was dried overnight at 60 °C under vacuum. 1H-NMR (400 MHz, D2O, 25 °C): δ = 4.60 (s, 3H, –OCH3), 6.84, 7.07, 7.14 and 7.27 (m, 1H each, H of anisole ring), 7.75 and 7.52 (d, 2H each, H of 4-carboxyphenyl ring), COOH (not observed).
For reaction 2 (2-bromopyridine + 4-carboxyphenylboronic acid) acetonitrile–water (7/3) was used as the eluent solution. Solvent was removed from the solution containing the product with a rotavapor and the orange solid obtained was dried at 60 °C under vacuum overnight. 1H-NMR (400 MHz, d6-DMSO, 25 °C): δ = 7.29 (m, 1H, Pyr), 7.86 (m, 2H, Pyr), 7.93 (m, 1H, Pyr), 7.78 and 7.69 (d, 2H each, H of 4-carboxyphenyl ring), 10.00 (br, 1H, COOH).
2.5.1.2. Quantification of the conversion rate. The technique employed to quantitatively analyze the conversion of 3-bromoanisole and to identify the reaction product was gas chromatography (GC) with FID detector (Varian CP-3370) using a 15 m CP-SIL-8 column provided by Scharlau. The kinetics or the conversion of the different reactions was determined by following the variation of the concentration of 3-bromoanisole in solution. To follow the changes of the halide concentration, an internal standard compound (1-octanol) was used. Comparing the areas relating to the signals of the halo compound and the internal standard, the concentration of the halide can be determined with accuracy. The chosen internal standard was 1-octanol (100 ppm), an inert compound with respect to reagents and products and that has a retention time different from the others compounds. The solution samples to be analyzed were diluted in the ratio 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 in order to adjust the concentration of reactants and products to the range of linearity of the calibration curve. The calibration curve was prepared using concentration of halide in the range 0–500 ppm for both reactions. The temperature conditions were: injector temperature: 240 °C, detector temperature: 250 °C, oven temperature program 130 °C (10 min); from 130 °C to 210 °C (with a ramp of 20 °C min−1) and 210 °C (10 min). Operating under these conditions the retention times of the chemical species in solution were, DMF: 3.5 min, 1-octanol: 6.5 min, 2-bromopyridine: 7.8 min, 3-bromoanisole: 12.0 min, 4-carboxyphenylboronic acid: 16.7 min, product of the first reaction: 20.0 min, product of the second reaction 19.6 min. The tests were carried out in triplicate, (the reported results are the average of those obtained). For an example of a chromatogram of the reaction mixture see Fig. S1 of the ESI.†
:10 dilutions of the samples were analyzed by GC.
:50 in order to adjust the concentration of reactants and products to the range of linearity of the calibration curve. The calibration curve was prepared using concentration of halide in the range 0–500 ppm for both reactions. The temperature conditions were: injector temperature: 240 °C, detector temperature: 250 °C, oven temperature program 130 °C (10 min); from 130 °C to 210 °C (with a ramp of 20 °C min−1) and 210 °C (10 min). Operating under these conditions the retention times of the chemical species in solution were, DMF: 3.5 min, 1-octanol: 6.5 min, 2-bromopyridine: 7.8 min, 3-bromoanisole: 12.0 min, 4-carboxyphenylboronic acid: 16.7 min, product of the first reaction: 20.0 min, product of the second reaction 19.6 min. The tests were carried out in triplicate, (the reported results are the average of those obtained). For an example of a chromatogram of the reaction mixture see Fig. S1 of the ESI.†
:10 dilutions of the samples were analyzed by GC.2.6. Cytotoxicity in vitro studies
The percentage of surviving cells was determined 96 h after the beginning of drug exposure. After 96 h treatment, the supernatant medium from the 96 well plates was eliminated and the cells were fixed with a 10% solution of trichloroacetic acid (TCA). After fixation, the cells were washed in a strip washer. The washing was carried out four times with water using alternate dispensing and aspiration procedures.
The plates were then dyed with 100 μL of 0.4% SRB for about 45 min. After dying, the plates were again washed to remove the dye with 1% acetic acid and dried to air overnight. 100 μL of 10 mM tris-hydroxymethylaminoethane (TRIS) was added to each well of the plate and absorbance was measured at 570 nm using a 96-well plate reader (Tecan Spectra, Crailsheim, Germany). The M50 value was estimated from the dose–response curves and was defined as the quantity (in μg mL−1) of the material at which 50% cell inhibition was observed.
3. Results and discussion
3.1. Synthesis and characterization of supported palladium nanoparticles
For SBA-15, the maximum percentage of incorporated palladium was 12.8 wt%; which was obtained when starting from theoretical 50 wt% Pd. The data from all the experiments showed a linear trend in the studied loading range (see Fig. S2 of ESI†). However, it seems unlikely that this linearity persists with higher percentages of initial palladium, even though; a maximum of saturation was not observed in the studied range (see Fig. S2† and Table 1).
For MSU-2, the maximum percentage of incorporated palladium was 4.46 wt% which was obtained when starting from a theoretical 20 wt% Pd. In this case, MSU-2 showed a non linear trend for the incorporation of palladium (see Fig. S3 of ESI† and Table 1), although, again, no saturation was found in the studied range. Compared to SBA-15, the amount of incorporated palladium was generally lower in MSU-2, probably due to the smaller average pore diameter of MSU-2.
 | ||
| Fig. 1 Nitrogen adsorption–desorption isotherms of SBA-15 and MSU-2. | ||
For the Pd-functionalized hybrid materials, all the N2 adsorption–desorption isotherms were very similar to those of their parent materials. As expected, all the Pd-functionalized materials exhibited slightly lower surface areas and lower average pore volume compared to their parent material, due to Pd deposition within the pores and/or on the surface of the materials. This probably leads to a slight blocking of the pore in detriment of the surface area and pore volume (see Table 2). This phenomenon was especially notable when increasing the Pd loading in samples SBA-15–Pd-50 and MSU-2–Pd-20. On the other hand, pore average diameter did not remarkably change after formation of the palladium nanoparticles.
| Material | Specific surface area (m2 g−1) | Pore size (Å) | Pore volume (cm3 g−1) |
|---|---|---|---|
| SBA-15 | 931 | 53.7 | 0.851 |
| SBA-15–Pd-1 | 785 | 55.9 | 0.785 |
| SBA-15–Pd-2 | 751 | 55.5 | 0.737 |
| SBA-15–Pd-5 | 763 | 54.8 | 0.738 |
| SBA-15–Pd-10 | 768 | 54.8 | 0.755 |
| SBA-15–Pd-20 | 714 | 55.4 | 0.708 |
| SBA-15–Pd-50 | 564 | 58.0 | 0.809 |
| MSU-2 | 1052 | 31.2 | 1.055 |
| MSU-2–Pd-1 | 970 | 32.5 | 0.990 |
| MSU-2–Pd-2 | 863 | 30.8 | 0.874 |
| MSU-2–Pd-5 | 979 | 32.3 | 0.989 |
| MSU-2–Pd-10 | 896 | 31.6 | 0.901 |
| MSU-2–Pd-15 | 934 | 32.2 | 0.943 |
| MSU-2–Pd-20 | 919 | 31.7 | 0.936 |
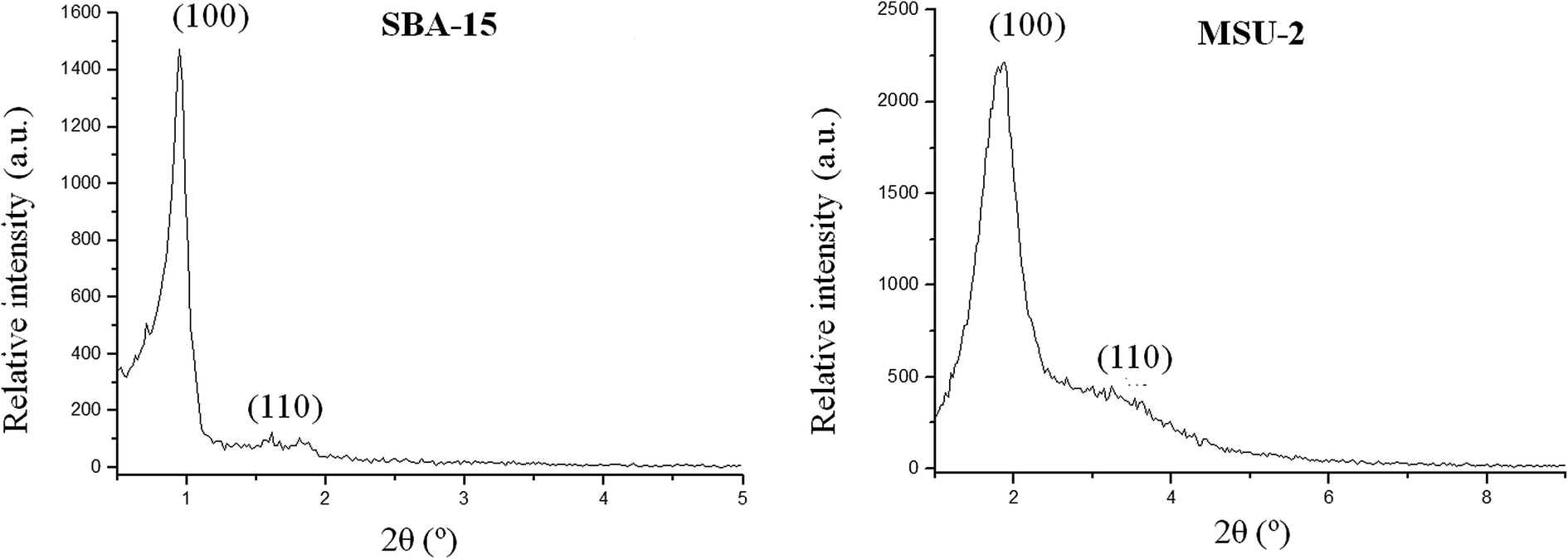 | ||
| Fig. 2 Low angle X-ray diffractograms of SBA-15 and MSU-2. | ||
The XDR reflections observed for the functionalized materials are similar to their non-functionalized analogues, with relatively small differences in the peak broadening and a slight decrease of their intensity due to a relatively small partial blocking of the dispersion centres as a consequence of the functionalization. This fact provides evidence that part of the functionalization process occurs inside the mesopore channels, since the attachment of metal nanoparticles here normally reduces the scattering power of the mesoporous silica walls. Analyzing the data of the interplanar distances of all silica-based mesoporous materials, it is possible to calculate the average wall thickness of the studied materials, using the following equation:
 | (1) |
The average wall thickness is 38.5 and 15.2 Å for unmodified SBA-15 and MSU-2, respectively. The wall thickness slightly increases after functionalization in the case of SBA-15 (with values from 38.6 to 40.2 Å), while it is apparently constant in the case of MSU-2 functionalized materials (with values from 14.6 to 15.9 Å). These results indicate that part of the Pd-nanoparticles may be located inside the pores of the corresponding materials.
For all the Pd-functionalized hybrid materials high angle X-ray diffractograms were recorded in order to observe the peaks associated to palladium nanoparticles (Fig. 3). Comparing the obtained diffractograms with others reported in the literature for Pd nanoparticles,2,32 the assignation of the corresponding Miller indices was possible. Thus, the most intense peaks at 2θ of 39.3, 45.4 and 67.5° were assigned to the (111), (200) and (220) planes, respectively, while the secondary peaks in intensity, at 80.3 and 84.6°, were assigned to the (311) and (222) planes, respectively.
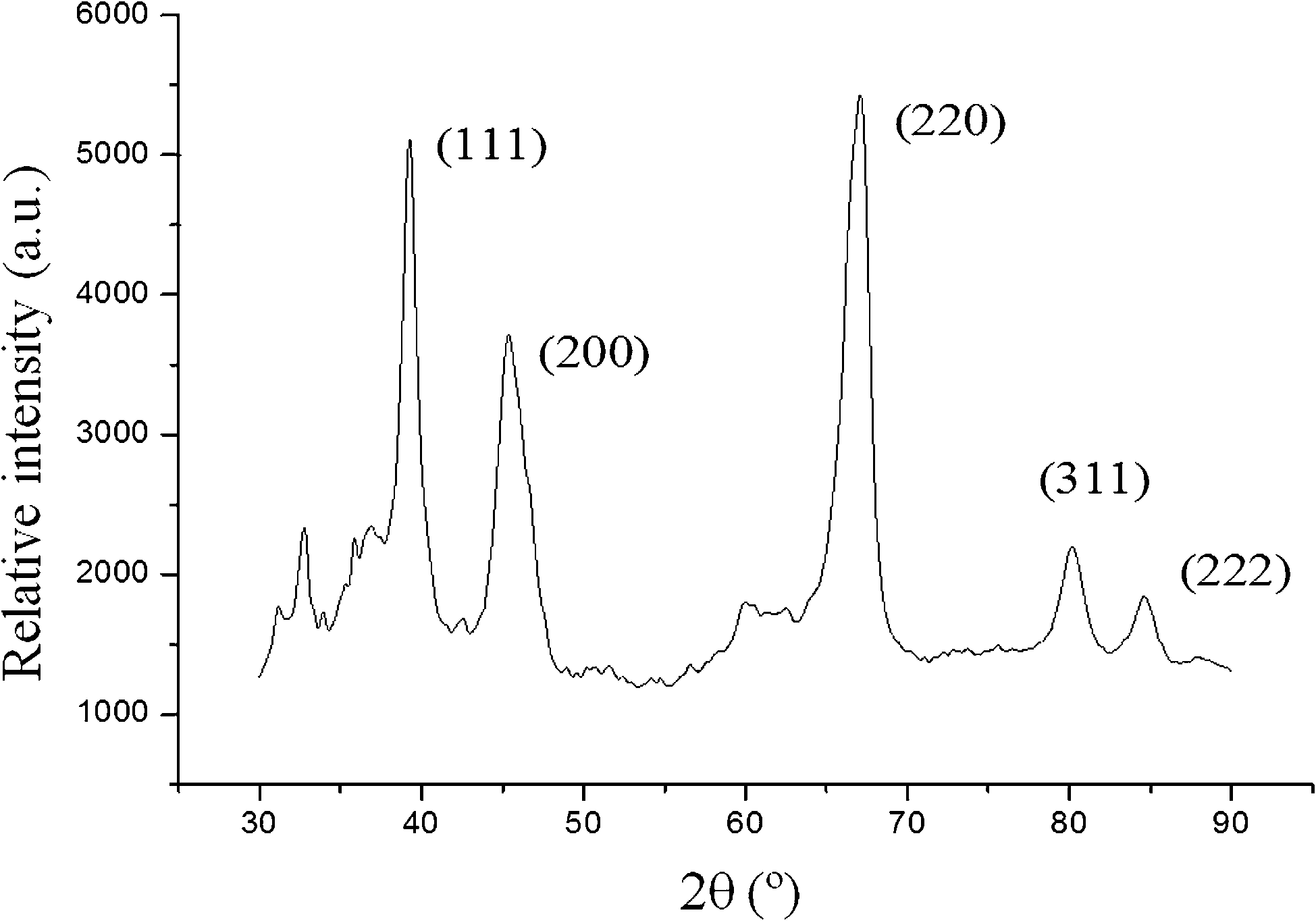 | ||
| Fig. 3 High angle X-ray diffractogram of SBA-15–Pd-50. | ||
 | ||
| Fig. 4 SEM images of (a) SBA-15 and (b) MSU-2. | ||
 | ||
| Fig. 5 TEM images of (a) and (b) SBA-15 and (c) and (d) MSU-2. | ||
For the Pd-functionalized materials (Fig. 6), one can easily detect the palladium nanoparticles as black dots either inside the pore of the mesoporous particles or impregnated on the external surface of the support. All hybrid materials show spherical palladium nanoparticles with diameters of 29 ± 9 nm for SBA-15-based materials (Fig. 6a) and 28 ± 5 nm for MSU-2-based materials (Fig. 6b) (for the particle size distribution see Fig. S7 and S8 of the ESI†). In all cases, the larger palladium particles appear to be clusters of very small nanoparticles.
 | ||
| Fig. 6 TEM images of Pd-functionalized materials (a) SBA-15–Pd-20 and (b) MSU-2–Pd-5. | ||
Reaction time has an influence on the morphology and location of palladium nanoparticles. The reaction of the formation of palladium nanoparticles was monitored by TEM. When using SBA-15, one can observe that after just 6 h of reaction (Fig. 7a) the main part of the palladium nanoparticles are located inside the pores showing a spherical or slightly elongated shape, where they grew following the directions of the channels. However, after 48 h of reaction, all the Pd-nanoparticles are impregnated on the outer surface of the mesoporous matrix (Fig. 6a).
 | ||
| Fig. 7 TEM images of Pd-functionalized materials (a) SBA-15–Pd-20 and (b) MSU-2–Pd-15 after 6 h of reaction. | ||
On the other hand, after just 6 h of reaction, the MSU-2-based materials did not show the palladium nanoparticles inside the non-regular pores of the mesoporous material (Fig. 7b), but always impregnated onto the mesoporous particle surface, observing almost no changes after 48 h of reaction (Fig. 6b).
As observed in the corresponding images (Fig. 6), palladium nanoparticles show a very high tendency to aggregate on the surface of the mesoporous materials (especially at higher loadings) resulting in a less homogeneous dispersion of the metal. The highly ordered structure of SBA-15 seems to promote a better incorporation of palladium nanoparticles within the mesoporous structure, as was indicated by the greater decrease in the specific surface area and pore volume, compared to MSU-2 (see data of N2 adsorption–desorption analyses in Table 2). However, applying longer reaction times to the non-ordered starting material MSU-2, leads to a better distribution of the Pd nanoparticles.
3.2. Catalytic tests
All the catalytic tests were carried out under previously reported experimental conditions,22 using a DMF–H2O (95/5) mixture as solvent, K2CO3 as base and measuring the final conversion 24 h after starting the reaction.
Table 3 shows the results obtained in the preliminary tests, for the reaction between 3-bromoanisole and 4-carboxyphenylboronic acid (Scheme 1) and Table 4 shows the results obtained in the catalytic reaction between 2-bromopyridine and 4-carboxyphenylboronic acid (Scheme 2).
| Catalytic test | Catalyst | Temperature (°C) | Br conversion (%) | Br conversion per Pd mg (%) |
|---|---|---|---|---|
| 1 | SBA-15–Pd-20 | 110 | 45 ± 1 | 14.7 ± 0.4 |
| 2 | SBA-15–Pd-20 | 70 | 51 ± 2 | 16.7 ± 0.7 |
| 3 | SBA-15–Pd-5 | 110 | 15 ± 1 | 25.0 ± 1.7 |
| 4 | SBA-15–Pd-5 | 70 | 29 ± 1 | 48.3 ± 1.7 |
| 5 | MSU-2–Pd-20 | 110 | 17 ± 2 | 7.6 ± 0.9 |
| 6 | MSU-2–Pd-20 | 70 | 19 ± 2 | 8.5 ± 0.9 |
| 7 | MSU-2–Pd-5 | 110 | 12 ± 2 | 34.3 ± 7.0 |
| 8 | MSU-2–Pd-5 | 70 | 15 ± 1 | 42.9 ± 3.5 |
| 9 | [Pd(PPh3)4] | 110 | 84 ± 3 | 168 ± 6 |
| 10 | [Pd(PPh3)4] | 70 | 92 ± 1 | 184 ± 2 |
| Catalytic test | Catalyst | Temperature (°C) | Br conversion (%) | Br conversion per Pd mg (%) |
|---|---|---|---|---|
| 11 | SBA-15–Pd-20 | 110 | 17 ± 2 | 5.6 ± 0.7 |
| 12 | SBA-15–Pd-20 | 70 | 33 ± 3 | 10.8 ± 1.0 |
| 13 | SBA-15–Pd-5 | 110 | 11 ± 1 | 18.3 ± 1.7 |
| 14 | SBA-15–Pd-5 | 70 | 24 ± 2 | 40.0 ± 3.4 |
| 15 | MSU-2–Pd-20 | 110 | 10 ± 1 | 4.5 ± 0.5 |
| 16 | MSU-2–Pd-20 | 70 | 28 ± 1 | 12.6 ± 0.5 |
| 17 | MSU-2–Pd-5 | 110 | 7 ± 1 | 20.0 ± 2.9 |
| 18 | MSU-2–Pd-5 | 70 | 14 ± 2 | 40.0 ± 5.8 |
| 19 | [Pd(PPh3)4] | 110 | 40 ± 1 | 80 ± 2 |
| 20 | [Pd(PPh3)4] | 70 | 63 ± 2 | 126 ± 4 |
It seems that the temperature has an influence on the conversion, observing, in all cases, higher catalytic activities when the temperature decreases from 110 to 70 °C. This phenomenon suggests that temperatures higher than 70 °C may lead to some changes in the physical structure either of the catalysts or of the reactants which may affect the catalytic activity.
In this context, material SBA-15–Pd-50 was analyzed by TEM after the reaction at 110 °C observing the formation of relatively big agglomerates of Pd nanoparticles (see Fig. S9 of ESI†). The agglomeration is normally favoured with an increase of the temperature and is associated with lower activity, thus giving a plausible explication for the catalytic results obtained. In addition, the higher catalytic activity of the reference compound [Pd(PPh3)4] at 70 °C compared to that of 110 °C may be explained by a possible deactivation of the catalyst due to the fact that at higher temperatures the formation of palladium nanoparticles may also occur and interfere with the catalytic reaction.33
A subsequent study of the halide conversion at different time periods in both reactions showed two different types of behavior. In catalytic test #9 between 3-bromoanisole and 4-carboxyphenylboronic acid, there is a rapid decrease of the concentration of the halide after the first 4 h followed by a slower decrease until the end of reaction time with a final conversion of 3-bromoanisole of 84% (see Fig. S10 of the ESI†). In the case of catalytic test #19 between 2-bromopyridine and 4-carboxyphenylboronic acid the concentration of the halide decreases more slowly and progressively during the reaction time, and does not seem to reach a minimum after 24 h (with a final conversion of 2-bromopyridine of 40%) (see Fig. S11 of the ESI†).
The differences in the conversion of both reactions seems to be justified by the fact that the meta-substituted arylbromides react faster than ortho-substituted derivatives (as is the case of 2-bromopyridine) due to steric factors. In addition, the inductive effect (−I) exerted by the methoxy group, which removes electron density, weakens the C–Br bond, increases its reactivity. The reactivity of 2-bromopyridine seems to be lowered also by the conjugative effect, due to the electron pair present at the nitrogen atom of the aromatic ring.
On analysis of the efficiency of the catalytic reactions regarding the halide conversion per mg of Pd in the mixture, one can see that the most effective catalysts are those with a lower Pd content. It seems that the Pd nanoparticles are more accessible in the case of those materials with a lower load, maybe due to a higher degree of agglomeration in materials with high palladium content. In addition, very similar efficiency was achieved for both MSU-2 or SBA-15-based materials (Tables 3 and 4).
In order to show the versatility of the synthesized hybrid materials in C–C coupling reactions, SBA-15–Pd-50 was also used as catalyst in the S–M reaction of 4-vinylboronic acid and 3-bromoanisole, observing a reasonable degree of halide conversion (41 ± 1%), which is lower to that described for the reaction with 4-carboxyphenylboronic acid using 15 mg (for further details see the ESI†). In addition, SBA-15–Pd-20 was tested as catalyst in the Sonogashira reaction of iodobenzene and phenylacetylene and a modest yield of 36% of diphenylacetylene was obtained (for further details see the ESI†).
The results of the tests using the studied materials as catalysts in the reaction between 3-bromoanisole and 4-carboxyphenylboronic acid are summarized in Table 5. Higher conversions were obtained using higher amounts of catalyst. This phenomenon may be due to influence of the Pd content which increases the halide conversion in this type of catalytic system.
| Catalytic test | Catalyst | Catalyst mass (mg) | Br conversion (%) | Br conversion per Pd mg (%) |
|---|---|---|---|---|
| 21 | SBA-15–Pd-50 | 15 | 66 ± 1 | 5.2 ± 0.1 |
| 22 | SBA-15–Pd-50 | 5 | 27 ± 2 | 2.1 ± 0.2 |
| 23 | MSU-2–Pd-15 | 15 | 39 ± 1 | 17.9 ± 0.5 |
| 24 | MSU-2–Pd-15 | 5 | 5 ± 1 | 2.3 ± 0.5 |
From these experiments using different quantities of catalyst, one observes, in both cases (SBA-15 and MSU-2), a higher catalytic efficiency (halide conversion per mg of Pd) using 15 mg of catalyst, than when using 5 mg of material. In addition, a slightly higher efficiency was achieved in these tests when using MSU-2-based materials compared to SBA-15 (Table 5). This probably arises from a better distribution of Pd nanoparticles in MSU-2–Pd-15 compared to that of the more Pd-loaded catalyst SBA-15–Pd-50 (see Section 3.2.1).
In the recycling tests a substantial loss of activity of the catalysts was observed after the first recycle test. A loss of activity of around 36% (from halide conversions of 63 to 39%) for SBA-15–Pd-50 and or ca. 35% (from halide conversions of 39 to 26%) for MSU-2–Pd-15 indicates the partial deactivation of the catalysts or leaching of the palladium nanoparticles after the first cycle. However, in the second and subsequent catalytic cycles the activity remained almost the same and did not show significant changes (Fig. 8). These results suggest a loss of activity by an initial leaching or deactivation of the supported catalyst that may affect only the non-impregnated palladium nanoparticles, which, because of their low size, are lost after the first catalytic cycle and subsequent filtration process. However, this does not happen after the second catalytic cycle because all the remaining Pd-nanoparticles are impregnated on the silica materials and do not leach after washing or filtration treatments. With this in mind, a TEM analysis of the materials used in the recyclability tests was carried out after the first catalytic cycle observing the formation of Pd nanoparticles agglomerates which are usually located on the external surface of the mesoporous materials (see Fig. S12 of the ESI†). This phenomenon may explain the lose of catalytic activity after the first catalytic cycle. For the second and subsequent catalytic cycles, there is no significant growth of the Pd palladium nanoparticle agglomerates which remain impregnated on the mesoporous silica Higher aggregates are probably not formed due to their lower mobility in suspension resulting from the larger particle size.
 | ||
| Fig. 8 Results of the recyclability tests using the catalysts SBA-15–Pd-50 and MSU-2–Pd-15. | ||
For the MSU-2 materials (non-ordered mesoporous silica), the decrease in the catalytic activity for the second and subsequent cycles is slightly lower to that of SBA-15. This might be due to a higher degree of dispersion of the nanoparticles in the MSU-2-based materials (with the same Pd load) and a lower tendency for the formation of agglomerates with respect to SBA-15, indicating a slight positive influence of the use of non-ordered mesoporous silica such as MSU-2.
In summary, these results show a good degree of stability of the studied catalysts in C–C coupling reactions although somewhat lower than some other similar systems described in the literature for the Heck reaction,34 and comparable to those described for Suzuki–Miyaura C–C coupling reactions.35,36
3.3. Preliminary cytotoxicity tests
Mesoporous silica-based materials functionalized with metal complexes have demonstrated very high cytotoxic activity and high potential for their application in chemotherapy.37–42 Bearing this in mind, a preliminary study of the cytotoxic activity of the Pd-functionalized materials SBA-15–Pd-50 and MSU-2–Pd-15, has been carried out in order to gain new insights on the cytotoxic properties of supported palladium nanoparticles and their potential as anticancer drugs.The materials have been analyzed in in vitro tests against anaplastic thyroid carcinoma (8505C), head and neck tumour (A253), lung carcinoma (A549) and colon carcinoma (DLD-1) cell lines. Cytotoxicity values, expressed as M50 values (the amount of material required to inhibit normal cell growth of the studied cell population by 50%), are given in Table 6. The studied materials SBA-15–Pd-50 and MSU-2–Pd-15 show a dose-dependent cytotoxicity (Fig. 9) against all the studied cancer cells. Taking into account that the unmodified mesoporous silica-based materials have not shown cytotoxic activity in previous studies reported by our group,37–42 it seems reasonable to assume that palladium nanoparticles play a crucial role in the cytotoxic action.
| Cell line | SBA-15–Pd-50 | MSU-2–Pd-15 |
|---|---|---|
| 8505C | 111 ± 20 | 417 ± 26 |
| A253 | 110 ± 16 | 284 ± 14 |
| A549 | 239 ± 20 | 553 ± 13 |
| A2780 | 207 ± 21 | 257 ± 15 |
| DLD-1 | 155 ± 6 | 241 ± 18 |
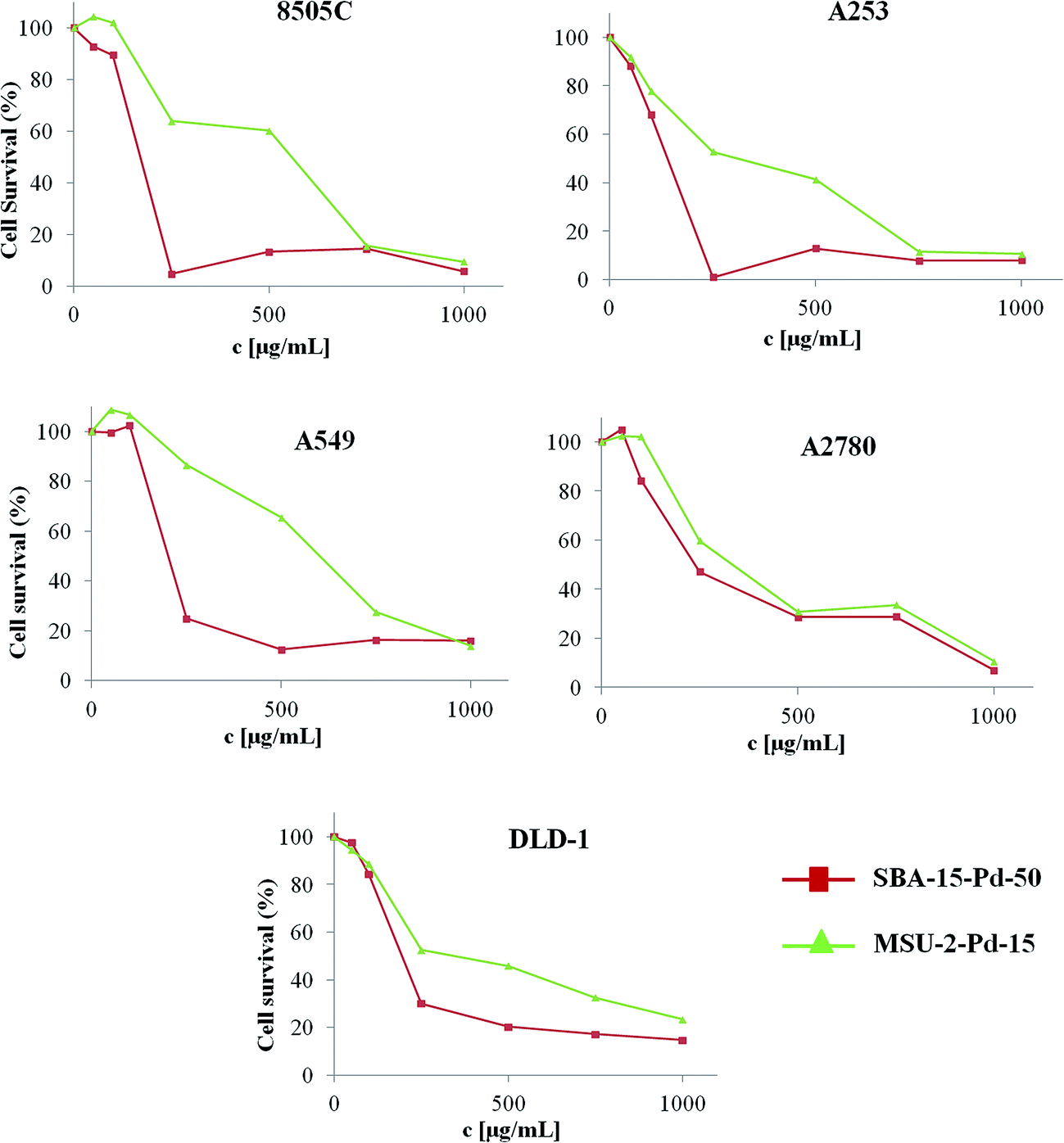 | ||
| Fig. 9 Cell survival vs. material concentration for different cancer cell lines (96 h). | ||
In addition, the fact that SBA-15–Pd-50 (with a Pd amount of 12.80 wt% and having M50 values from 110 ± 16 to 239 ± 20 μg mL−1) shows higher cytotoxic activity in all the studied cancer cell lines than MSU-2–Pd-15 (with a Pd amount of 2.18 wt% with M50 values from 241 ± 18 to 553 ± 13 μg mL−1), indicates again the influence of the palladium nanoparticles and that palladium content is essential to give a substantial increase in the cytotoxic activity.
The comparison of the cytotoxic activity of the studied materials with other mesoporous silica-based materials functionalized with metal complexes, shows the M50 values of SBA-15–Pd-50 are one of the lowest of all the studied systems, indicating their relatively high cytotoxic activity. In this context, only the recently reported tin-41 and titanocene-functionalized materials42 have shown higher cytotoxic activity in cancer cell lines.
Interestingly, both materials reported here, SBA-15–Pd-50 and MSU-2–Pd-15, have shown a lower cytotoxicity against A549 cells compared with the other tested cell lines. This phenomenon led us to believe that there might be some differences in the mechanism associated to the cell death promoted by these hybrid materials in the cancer cell lines. However, caution must be taken when analyzing the tendencies in the cytotoxic nature of these materials as many factors such as, the quantity of material used, palladium content, studied cell type and mesoporous silica type, come into play.
4. Conclusions
Supported palladium nanoparticles have been synthesized using SBA-15 and for the first time using MSU-2 as supports and [PdCl2(cod)] as a palladium precursor in a simple reaction which does not need the use of functionalized silica-based nanostructured materials. The functionalization reactions show different tendencies for the incorporation of the metal nanoparticles depending on the supporting material with a maximum load of Pd of 12.80 wt% on SBA-15 and 4.46 wt% on MSU-2, observing that systems with a disordered arrangement of the pores (MSU-2) gave lower Pd conversion to nanoparticles. The hybrid materials have been used as catalysts for the Suzuki–Miyaura (SM) coupling reaction between 3-bromoanisole and 4-carboxyphenylboronic acid and the reaction between 2-bromopyridine and 4-carboxyphenylboronic acid. Maintaining the same reaction conditions, a higher catalytic activity was observed at 70 °C compared to 110 °C. This is probably due to the formation of bigger agglomerates of palladium nanoparticles at higher temperatures which partially deactivate the hybrid catalysts, as was deduced from the TEM analysis of the resulting materials after the catalytic reaction. The reaction between 3-bromoanisole and 4-carboxyphenylboronic acid catalyzed by the hybrid materials and the reference catalyst [Pd(PPh3)4] is faster with both heterogeneous and homogeneous catalysts (needing only 24 h for the maximum conversion) than that of 2-bromopyridine and 4-carboxyphenylboronic acid (which needs around 48 h) and this may be due either to steric and/or electronic factors. In addition, the results show that the most effective catalysts are those with a lower Pd content. It seems that the Pd nanoparticles are more accessible in the case of those materials with a lower load, that have a better distribution of the palladium nanoparticles and/or a higher degree of agglomeration in materials with respect to materials with high palladium content. However, very similar efficiency was achieved for both MSU-2- and SBA-15-based materials in the studied catalytic tests. Additionally, in the reaction between 3-bromoanisole and 4-carboxyphenylboronic acid catalyzed by SBA-15–Pd-50 or MSU-2–Pd-15, conversion of the halide compound was quantified using 15 mg or 5 mg of fresh catalyst observing that the highest conversions were obtained when using 15 mg indicating the influence of the quantity of palladium on the final catalytic activity and a slightly higher catalytic efficiency was observed when using 15 mg. A recyclability study using SBA-15–Pd-50 and MSU-2–Pd-15 in up to six catalytic cycles was carried out observing that the conversion of the halide is reduced after the first cycle, however, it remains nearly constant for the other five cycles indicating a reasonable degree of recyclability of the studied materials. This phenomenon is probably due to the significant agglomeration of Pd-particles after the first catalytic cycle. This is not as dramatic in the second and subsequent catalytic cycles because the bigger Pd nanoparticles remain impregnated on the mesoporous silica without forming higher aggregates due to their lower mobility in suspension associated to their higher particle size.Hybrid materials SBA-15–Pd-50 and MSU-2–Pd-15 have been tested in vitro against human cancer cells observing a dose dependent cytotoxicity in all the studied cancer cell lines and a higher cytotoxic effect of SBA-15–Pd-50 compared to MSU-2–Pd-15. According to our experiments it seems that the cytotoxic nature of these materials depends on the quantity of material used, palladium content, studied cell type and mesoporous silica type. To the best of our knowledge, this is the first report about the cytotoxic activity of supported palladium nanoparticles and further study on the mechanistic aspects of the cell death induction promoted by these materials needs to be carried out in order to determine if these palladium nanoparticle functionalized materials are potential candidates for therapeutic treatments.
Acknowledgements
We gratefully acknowledge financial support from the Ministerio de Economía y Competitividad, Spain (Grant no. CTQ2012-30762). A. Benedetti and D. Pérez are gratefully acknowledged for helpful discussion.References
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- V. Budarin, J. H. Clark, R. Luque, J. Macquarrie, J. Robin and R. White, Green Chem., 2008, 10, 382–387 RSC.
- V. Mody, R. Siwale, A. Singh and H. R. Mody, J. Pharm. BioAllied Sci., 2010, 2, 282–289 CrossRef CAS PubMed.
- Nanoparticles and Catalysis, ed. D. Astruc, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008 Search PubMed.
- W. A. Lopes and H. M. Jaeger, Nature, 2001, 414, 735–738 CrossRef CAS PubMed.
- J. S. Aaron, J. Oh, T. A. Larson, S. Kumar, T. E. Milner and K. V. Sokolov, Opt. Express, 2006, 14, 12930–12943 CrossRef CAS PubMed.
- A. B. Laursen, K. T. Højholt, L. F. Lundegaard, S. B. Simonsen, S. Helveg, F. Schüth, M. Paul, J.-D. Grunwaldt, S. Kegnæs, C. H. Christensen and K. Egeblad, Angew. Chem., Int. Ed., 2010, 49, 3504–3507 CrossRef CAS PubMed.
- I. Slowing, J. L. Vivero-Escoto, B. G. Trewyn and V. S.-Y. Lin, J. Mater. Chem., 2010, 20, 7924–7937 RSC.
- V. Barau, A. Budarin, R. Caragheorgheopol, D. J. Luque, A. Macquarrie, V. S. Prelle, M. Teodorescu and A. Zaharescu, Catal. Lett., 2008, 124, 204–214 CrossRef.
- J. He, I. Ichinose, T. Kunitake and A. Nakao, Langmuir, 2002, 18, 10005–10010 CrossRef CAS.
- L. Guczi, A. Beck, A. Horváth and D. Horváth, Top. Catal., 2002, 19, 157–163 CrossRef CAS.
- H. Ye, R. W. J. Scott and R. M. Crooks, Langmuir, 2004, 20, 2915–2920 CrossRef CAS PubMed.
- A. Troupis, E. Gkika, A. Hiskia and E. Papaconstantinou, C. R. Chim., 2006, 9, 851–857 CrossRef CAS.
- Á. Mastalir, B. Rác, Z. Király and Á. Molnár, J. Mol. Catal. A: Chem., 2007, 264, 170–178 CrossRef.
- K. Liu, Z. X. Chen, Z. Q. Hou, Y. Y. Wang and L. Y. Dai, Catal. Lett., 2014, 144, 935–942 CrossRef CAS.
- P. Li, H. Liu, Y. Yu, C. Y. Cao and W. G. Song, Chem.–Asian J., 2013, 8, 2459–2465 CrossRef CAS PubMed.
- C. M. A. Parlett, D. W. Bruce, N. S. Hondow, M. A. Newton, M. A. A. F. Lee and K. Wilson, ChemCatChem, 2013, 5, 939–950 CrossRef CAS.
- I. Yuranov, P. Moeckli, E. Suvorova, P. Buffat, L. Kiwi-Minsker and A. Renkena, J. Mol. Catal. A: Chem., 2003, 192, 239–251 CrossRef CAS.
- S. MacQuarrie, B. Nohair, J. H. Horton, S. Kaliaguine and C. M. Crudden, J. Phys. Chem. C, 2010, 114, 57–64 CAS.
- C. Y. Ma, B. J. Dou, J. J. Li, J. Cheng, Q. Hu, Z. P. Hao and S. Z. Qiao, Appl. Catal., B, 2009, 92, 202–208 CrossRef CAS.
- R. Xing, Y. Liu, H. Wu, X. Li, M. He and P. Wu, Chem. Commun., 2008, 6297–6299 RSC.
- M. Trilla, G. Borja, R. Pleixats, M. W. C. Man, C. Bied and J. J. E. Moreau, Adv. Synth. Catal., 2008, 350, 2566–2574 CrossRef CAS.
- A. Speranza, K. Leopold, M. Maier, A. R. Taddei and V. Scoccianti, Environ. Pollut., 2010, 158, 873–882 CrossRef CAS PubMed.
- C. P. Adams, K. A. Walker, S. O. Obare and K. M. Docherty, PLoS One, 2014, 9, e85981 Search PubMed.
- D. De Stefano, R. Carnuccio and M. C. Maiuri, J. Drug Delivery, 2012, 167896 Search PubMed.
- C. Petrarca, E. Clemente, L. Di Giampaolo, R. Mariani-Costantini, K. Leopold, R. Schindl, L. V. Lotti, R. Mangifesta, E. Sabbioni, Q. Niu, G. Bernardini and M. Di Gioacchino, J. Immunol. Res., 2014, 295092 Search PubMed.
- M. Horie, K. Fujita, H. Kato and S. Endoh, Metallomics, 2012, 4, 350–360 RSC.
- D. Zhao, Q. Huo, J. Feng, B. Chmelka and G. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- D. Pérez-Quintanilla, A. Sánchez, I. Hierro, M. Fajardo and I. Sierra, J. Nanosci. Nanotechnol., 2009, 9, 4901–4909 CrossRef.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- S. Navaladian, B. Viswanathan, T. K. Varadarajan and R. P. Viswanath, Nanoscale Res. Lett., 2009, 4, 181–186 CrossRef CAS PubMed.
- R. Molina, S. Gómez-Ruiz, F. Montilla, A. Salinas-Castillo, S. Fernández-Arroyo, M. M. Ramos, V. Micol and R. Mallavia, Macromolecules, 2009, 42, 5471–5477 CrossRef CAS.
- M. Arpad, Chem. Rev., 2011, 111, 2251–2320 CrossRef PubMed.
- G. H. Zhang, P. Y. Wang and X. F. Wei, Catal. Lett., 2013, 143, 1188–1194 CrossRef CAS.
- B. Basu and S. Paul, Appl. Organomet. Chem., 2013, 27, 588–594 CAS.
- D. Perez-Quintanilla, S. Gómez-Ruiz, Ž. Žizak, I. Sierra, S. Prashar, I. del Hierro, M. Fajardo, Z. D. Juranić and G. N. Kaluđerović, Chem.–Eur. J., 2010, 12, 5588–5597 Search PubMed.
- G. N. Kaluđerović, D. Perez-Quintanilla, I. Sierra, S. Prashar, I. del Hierro, Ž. Žizak, Z. D. Juranić, M. Fajardo and S. Gómez-Ruiz, J. Mater. Chem., 2010, 20, 806–814 RSC.
- G. N. Kaluđerović, D. Perez-Quintanilla, Ž. Žizak, Z. D. Juranić and S. Gómez-Ruiz, Dalton Trans., 2010, 39, 2597–2608 RSC.
- A. García-Peñas, S. Gómez-Ruiz, D. Perez-Quintanilla, R. Paschke, I. Sierra, S. Prashar, I. del Hierro and G. N. Kaluđerović, J. Inorg. Biochem., 2012, 106, 100–110 CrossRef PubMed.
- M. Z. Bulatović, D. Maksimović-Ivanić, C. Bensing, S. Gómez-Ruiz, D. Steinborn, H. Schmidt, M. Mojić, A. Korać, I. Golić, D. Pérez-Quintanilla, M. Momčilović, S. Mijatović and G. N. Kaluđerović, Angew. Chem., Int. Ed., 2014, 53, 5982–5987 CrossRef PubMed.
- J. Ceballos-Torres, P. Virag, M. Cenariu, S. Prashar, M. Fajardo, E. Fischer-Fodor and S. Gómez-Ruiz, Chem.–Eur. J., 2014, 20, 10811–10828 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11759j |
| This journal is © The Royal Society of Chemistry 2014 |