
Design and synthesis of novel carbazolo–thiazoles as potential anti-mycobacterial agents using a molecular hybridization approach†
Abstract
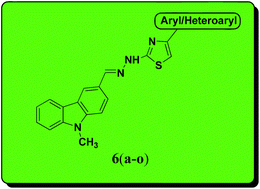
Various substituted carbazolo–thiazoles (compounds 6a–6o) were synthesized in good yields using a molecular hybridization approach. The synthesized compounds were evaluated for their in vitro anti-tubercular activity against Mycobacterium tuberculosis H37Rv strain at the National Institute of Allergy and Infectious Diseases (Bethesda, MD, USA). Among the tested series, compound 6c (minimum inhibitory concentration 21 μM) showed the most promising anti-mycobacterial activity. Brief structure–activity relationship studies showed that the electron-donating groups (OCH3 and OH), particularly on the phenyl ring of the thiazole motif, had a positive correlation with the anti-mycobacterial activity. In addition, they displayed low cytotoxicity against a mammalian Vero cell line using the MTT assay, thereby having a high therapeutic index. This study shows the importance of molecular hybridization and the scope for the development of carbazole–thiazole compounds as potential anti-mycobacterial agents.
 Please wait while we load your content...
Please wait while we load your content...

