
Antioxidant properties of several coumarin–chalcone hybrids from theoretical insights†
Gloria Mazzone*ab,
Naim Malajac,
Annia Galanod,
Nino Russoa and
Marirosa Toscanoa
aDipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, I-87036 Arcavacata di Rende, Italy. E-mail: gloria.mazzone@unical.it
bDipartimento di ingegneria Informatica, Università della Calabria, Modellistica, Elettronica e Sistemistica, I-87036 Arcavacata di Rende, Italy
cDipartimento di Scienze Ambiente e Territorio e Scienze della Terra, Università di Milano-Bicocca, Via Pietro Bucci, 87036, Rende, Italy
dDepartamento de Quimica, Division de Ciencias Basicas e Ingenieria, Universidad Autonoma Metropolitana-Iztapalapa, Av. San Rafael Atlixco No. 186, Col. Vicentina, CP 09340, Mexico, D.F., Mexico
First published on 19th November 2014
Abstract
Density functional theory (DFT) and time-dependent formulation of DFT (TDDFT) have been employed to elucidate the structural characteristics, the antioxidant ability, and the UV-Vis absorption properties of a series of coumarin–chalcone derivatives recently synthesized. In addition, to investigate the role of adjacent hydroxyl groups on the antioxidant properties, five additional hybrids were designed and considered in this study. Different antioxidant mechanisms have been investigated. They are hydrogen atom transfer (HAT), electron transfer followed by proton transfer (SET-PT), and sequential proton loss electron transfer (SPLET). Based on the obtained results, the HAT mechanism is proposed as the most important one for the antioxidant protection exerted by this class of compounds. The UV spectra of coumarin–chalcone hybrids are characterized by a band in the region between 300 and 450 nm arising from different electronic transitions. Our investigation confirms the antioxidant properties of these hybrids, and shows that poly-substitution of ring A enhances the antioxidant power of this class of compounds. One of the derivatives, designed in the present work, the 5,6,8-trihydroxy-7-methyl-3-(3′,4′-dihydroxybenzoyl) coumarin, seems to be the most promising candidate as an antioxidant. Accordingly, our calculations strongly encourage the synthesis of coumarin–chalcone hybrids as an important strategy to develop novel compounds with improved antioxidant properties.
Introduction
The increasing evidence supporting the beneficial effects of diverse natural compounds, occurring in plant tissues, have motivated the research community to characterize their chemical and biological properties. Most of the active natural compounds are plant secondary metabolites. Coumarins (2H-1-benzopyran-2-ones) and chalcones (1,3-diphenyl-2-propen-1-ones) are among such natural compounds that are distributed throughout all parts of the plant, including fruits, seeds, leaves, and roots. Several members of these two families of compounds have been associated with numerous biological activities,1–6 amongst which the antioxidant capacity, that expresses the ability to deactivate free radicals, is probably the most important one.5,7–11Free radicals are unstable chemical species, with unpaired electrons, that are usually highly reactive toward other species. They can be classified as reactive oxygen species (ROS), reactive nitrogen species (RNS) and reactive sulphur species (RSS). Even though free radicals are mainly produced through regular metabolic routes, there are also some external factors that promote their production, including smoking, environmental pollutants, radiation and drugs, amongst others.9,12 In healthy organisms, there is a delicate balance between the production and the removal of free radicals, which guarantees that they remain in adequate concentrations. However, when this balance is broken these reactive species start producing chemical damages to proteins, lipids, DNA, RNA and sugars generating the so called oxidative stress of body cells.9,13 These processes have been associated with several diseases including cardiovascular, liver, neurological, and renal disorders, as well as cancer, auto-immune deficiency, and degenerative disorders associated with aging, diabetes, obesity, autism, Alzheimer's, Parkinson's and Huntington's diseases.9,12,14 Although humans have developed different endogenous defence mechanisms to protect cells from the excess of free radicals and to avoid the oxidative stress, often these mechanisms are not enough. Therefore, to increase protection from oxidative damage, dietary supplements with antioxidants are recommended as a way to maintain the concentration of free radicals as low as possible.15
There are three particularly important mechanisms for the free radical scavenging activity of phenolic compounds (ArOH). The first one is the hydrogen atom transfer (HAT, reaction (1)) which occurs in one single step. In this mechanism the free radical is responsible for the hydrogen abstraction from the antioxidant molecule. Therefore, the bond dissociation enthalpy (BDE) referred to the O–H bonds of the phenolic antioxidants indicates its ability to transfer a hydrogen atom to a free radical. Compounds with high antioxidant activity typically show low BDE values indicating an easier O–H dissociation and consequently a greater interaction with free radicals.
| ArOH + ROO˙ → ArO˙ + ROOH | (1) |
The second mechanism, single-electron transfer followed by proton transfer (SET-PT), takes place in two steps: the radical cation ArOH+˙ is formed first via electron transfer from the antioxidant to the free radical (reaction (2.1)), and then it deprotonates yielding the ArO˙ radical (reaction (2.2)), followed by ROOH formation (reaction (2.3)). Therefore, the adiabatic ionization potentials (IP) and O–H proton dissociation enthalpies (PDE) provide information about the energetic of this process. Low IP values indicate a greater tendency to form a superoxide radical anion enhancing the antioxidant activity.
| ArOH + ROO˙ → ArOH+˙ + ROO− | (2.1) |
| ArOH+˙ → ArO˙ + H+ | (2.2) |
| ROO− + H+ → ROOH | (2.3) |
The sequential proton loss electron transfer (SPLET), also takes place through two consecutive steps. In this case the first one is the deprotonation of the phenolic compound. Thus, the proton affinity (PA) of the phenoxide anion (ArO−) is adequate to assess the viability of this step (reaction (3.1)). On the other hand, the subsequent electron transfer from the phenoxide anion to ROO˙, to form the phenoxyl radical, is well depicted by the electron transfer enthalpy (ETE).
| ArOH → ArO− + H+ | (3.1) |
| ArO− + ROO˙ → ArO˙ + ROO− | (3.2) |
| ROO− + H+ → ROOH | (3.3) |
The final products yielded by these three mechanisms (HAT, SET-PT, and SPLET) are exactly the same: ArO˙ and ROOH.
For a particular compound to be a good antioxidant, the new radical species formed after the scavenging reaction should be more stable and innocuous for cells than the initial free radical. Coumarins and chalcones, as well as many phenolic compounds, found in edible parts of fruits and vegetables, fulfill well this requirement. Additionally, in the last few years, molecules belonging to different classes of compounds have been synthetically combined in order to obtain hybrids with enhanced biological activities.16–19 This hybridization approach inspired Vazquez-Rodriguez and collaborators20 to use coumarin and chalcone moieties as starting molecules to synthesize five coumarin–chalcone hybrids (Scheme 1, compounds 1–5) in order to obtain compounds with improved antioxidant and trypanocidal activity against Trypanosoma cruzi, the parasite responsible for Chagas disease. The authors applied the cyclic voltammetry, the oxygen radical absorbance capacity (ORAC) and the electron spin resonance (ESR) approaches to evaluate the antioxidant activity of the synthesized hybrids. They found that the presence of bromine and hydroxyl groups in the coumarin moiety (compound 4 and 5) leads to an improvement in the antioxidant activity.
 | ||
| Scheme 1 Schematic drawing of all the investigated compounds. | ||
In order to rationalize the experimental results obtained by Vazquez-Rodriguez et al.,20 regarding the antioxidant activity, we report here a DFT-based computational investigation about the mechanisms by which the synthesized coumarin–chalcone hybrids exert their action against free radicals. In addition, we included in the present work the theoretical investigation of five similar coumarin–chalcone derivatives (Scheme 1, compounds 6–10) designed by us.
The relationship between the experimental radical scavenging activity of several coumarin–chalcone derivatives and the calculated reaction enthalpies (BDE, IP, PDE, PA and ETE) is analyzed. The energies of frontier orbitals, as well as a comparison between the measured first oxidation potentials and those obtained from the calculations are provided. Details about the conformational and electronic features of the five coumarin–chalcones under investigation, the effect of the position of substituents on their antioxidant ability are also presented.
Computational details
The geometries of all the investigated phenolic compounds, including radicals, radical cations, and anions, have been fully optimized in methanol media, employing the hybrid functional B3LYP, which is constructed from the exchange functional proposed by Becke (B3) in combination with the Lee, Yang, and Parr (LYP) correlation functional.21,22 All the electronic calculations were performed using the Gaussian 09 computational package.23For all the atoms involved, the geometry optimizations were performed using the 6-31+G(d,p) basis set. The solvation effects were computed by using the Conductor Polarizable Continuum Model (CPCM).24 The UFF set of radii was used to build-up the cavity. The dielectric constant 32.6 was used in order to simulate the solvent (methanol). Harmonic vibrational frequency calculations have been performed to confirm that all the optimized structures as actual minima. Unrestricted calculations were used for the open-shell systems, such as radicals and radical cation species. No spin contamination was found, being the 〈S2〉 value about 0.750 in all cases. Final energies were calculated by performing single-point calculations on the optimized geometries at the same level of theory and employing a larger standard basis set, 6-311++G(3df,2p), for all the atoms. Computations of single-point spin densities were performed using the above mentioned protocol for the most stable open shell species. This computational protocol has already been successfully applied to investigate the antioxidant properties of a large series of polyphenols.25–31
BDE, IP, PDE, PA and ETE were calculated, using methanol as a solvent, at 298.15 K, according to the following expressions:
| BDE = H(ArO˙) + H(H˙) − H(ArOH) |
| IP = H(ArOH˙+) + H(e−) − H(ArOH) |
| PDE = H(ArO˙) + H(H+) − H(ArOH˙+) |
| PA = H(ArO−) + H(H+) − H(ArOH) |
| ETE = H(ArO˙) + H(e−) − H(ArO−) |
The H(H+) and H(e−) enthalpy values were taken from experimental measurements.32 However, theoretical determinations of these quantities are available in a previous DFT study on some phenolic derivatives.33
The standard redox potential is related to the free energy difference of the reaction through the following thermodynamic relation:
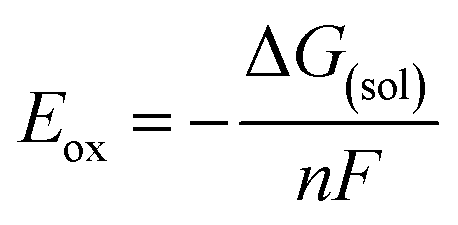
| ArOH(sol) → ArOH(sol)+˙ + e− | (1) |
Using the Born–Haber thermodynamic cycle outlined in the Scheme S1 reported in the ESI,† ΔG(sol) for a one-electron oxidation of a given phenolic species is defined as:
| ΔG(sol) = ΔG(gas) + ΔG(solv,ArOH+˙) − ΔG(solv,ArOH) |
Since in the reference experimental paper the authors performed cyclic voltammetry measurements using Ag/AgCl (0.222 V) as a reference electrode, the calculated value for the oxidation potential (Eox) is relative to the reduction potential of the Ag/AgCl electrode.
Finally, absorption spectra were computed as vertical electronic excitations from the minima of the ground-state structures by using time-dependent density functional response theory34 as implemented in the Gaussian 09 code. These calculations were carried out in methanol medium, using the standard 6-31+G(d,p) basis set and B3LYP, PBE0 and wB97XD exchange-correlation functionals.
Results and discussion
Antioxidants (ArOH) prevent extensive peroxidation of lipids, and other essential biomolecules, by transferring their phenolic H atoms or a single electron to a free radical at a faster rate than that of chain propagation. In this kind of reactions, the chemical structure of antioxidants is a key factor for their radical scavenging ability. Phenolic species present, as their main structural characteristic, one or more hydroxyl groups attached to aromatic rings. The concomitant presence of more than one hydroxyl groups, as well as the position of such groups on the rings, is crucial for a good antioxidant activity. In fact, these are the key features for the structure–activity relationships of these compounds. Therefore, detailed knowledge of the structural and electronic characteristics of phenols is of crucial importance in elucidating their scavenging behaviour.The coumarin–chalcone derivatives contain multiple OH groups (Scheme 1), which can yield different radicals depending on the group involved in the H transfer reaction.
Molecular geometries and radicals stability
The optimized structures of all the investigated compounds are reported in Fig. S2 of the ESI,† while selected geometrical parameters, and relative enthalpies, of their radicals (formed via HAT) are reported in Table 1. In this table the lowest BDE is taken as the reference value (0.00 kcal mol−1) in order to highlight the energy difference between the most stable radical, of each compound, and the other ones. In addition, the spin densities of the most stable radicals are reported in Fig. S3 of the ESI† to allow a better visualization of the delocalization of the unpaired electron.| Radicals | Compounds, ΔE | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| 3′-OH | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 7.97 | 1.43 | 4.07 | 11.94 | 6.51 |
| 4′-OH | 0.12 | 0.11 | 0.11 | 0.08 | 0.10 | 7.85 | 1.42 | 4.08 | 11.83 | 6.40 |
| 5-OH | 6.12 | 0.00 | 0.00 | 0.00 | 0.00 | |||||
| 6-OH | 4.85 | 1.05 | 1.31 | 3.83 | ||||||
| 7-OH | 7.50 | 3.45 | 1.61 | |||||||
| 8-OH | 5.18 | 0.69 | 0.00 | 6.68 | 2.65 | |||||
| Angles |
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| φ1 (C3–Cα–C1′–C2′) | −17.7 | −17.9 | −16.8 | −22.7 | −15.7 | −18.4 | −20.4 | −19.0 | −18.1 | −20.7 |
| φ2 (C2–C3–Cα–O) | 126.1 | 126.0 | 124.5 | 134.1 | 123.3 | 127.8 | 131.4 | 129.8 | 127.7 | 131.6 |
| θ1 (C10–C5–R1) | 119.0 | 118.5 | 120.1 | 116.4 | 120.0 | 119.1 | 119.3 | 119.0 | 123.4 | 121.9 |
| θ2 (C5–C6–R2) | 120.1 | 121.6 | 117.6 | 120.4 | 119.4 | 117.6 | 121.3 | 115.0 | 120.1 | 120.3 |
| θ3 (C6–C7–R3) | 119.8 | 119.1 | 119.9 | 121.6 | 121.3 | 117.0 | 119.1 | 119.4 | 120.0 | 120.3 |
| θ4 (C7–C8–R4) | 121.7 | 121.5 | 121.0 | 121.0 | 119.4 | 117.1 | 117.5 | 121.2 | 119.5 | 120.0 |
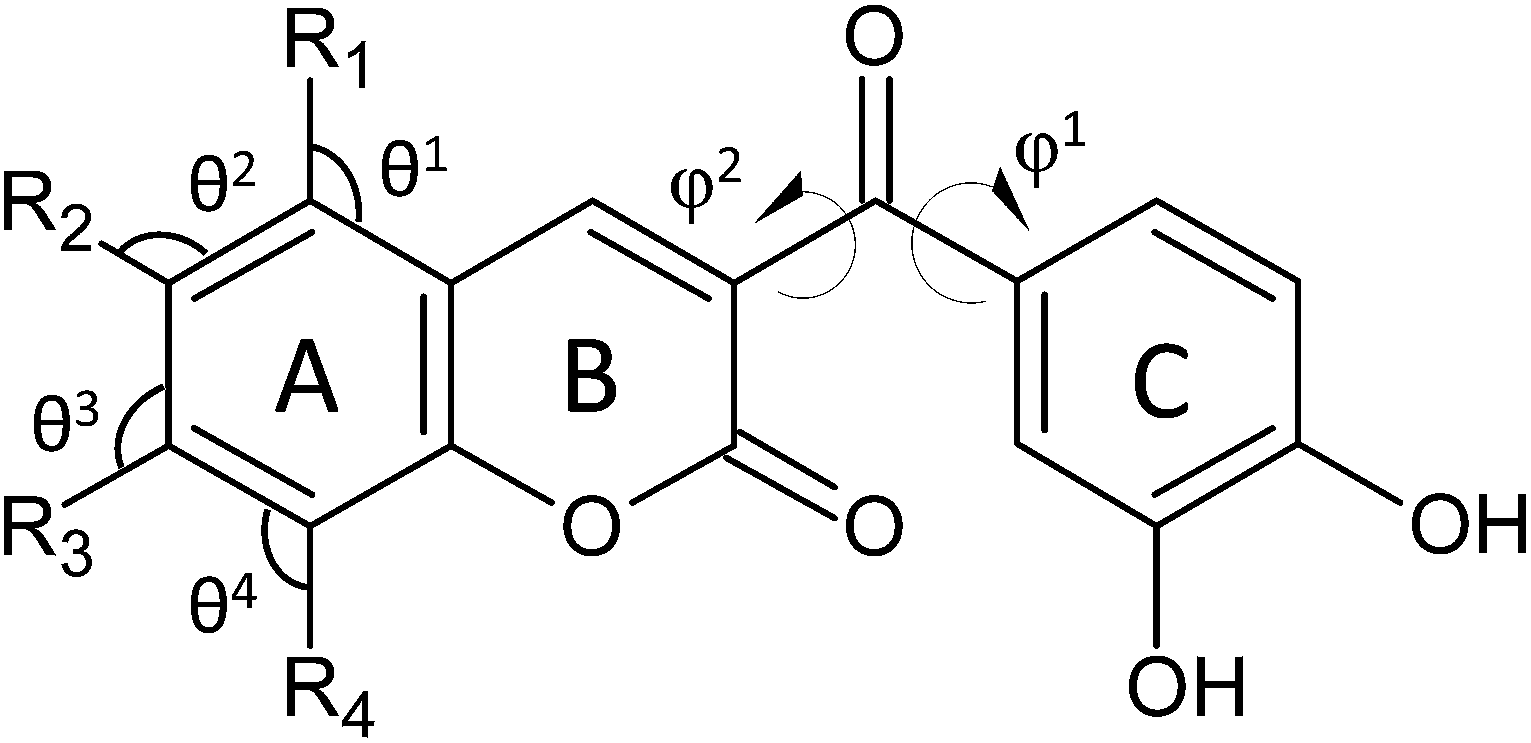
Compound 1 is, structurally, the simplest one with only two OH groups, located on the catechol part (ring C) of the coumarin–chalcone hybrid. This characteristic catechol moiety is common and remain unchanged for all the studied compounds. The coumarinic portion is completely planar, while the C ring slightly deviates from planarity, and it is characterized by a dihedral angle φ1 = −17.7°, established by the carbon atoms in positions 3, α, 1′ and 2′, and a dihedral angle φ2 (C2–C3–Cα–O) of 126.1°. Results reported in Table 1 indicate that the radicalization of both 3′- and 4′-hydroxyl groups leads to radical species equally stable. Compound 2 is structurally very similar to compound 1, with the difference that it presents a methyl group on the C6 position. The presence of this group on the coumarinic moiety does not affect neither the geometric structure nor the radicals' stability. In fact, both dihedral angles φ1 and φ2 are the same as those found in compound 1 and the energy difference between the two possible radicals is very small, indicating that C3′–OH and C4′–OH radicals have essentially the same stability. In addition the insertion of a third hydroxyl group in compound 3, does not introduce significant changes in the optimized geometry (see angles in Table 1). The same is found for the stability differences among radicals generated by the H-abstraction from OH groups on the catechol portion of the molecule, which result equally stables. The radical 6-OH, instead, is less stable than the 3′-OH one by 4.85 kcal mol−1. In compound 4, the concomitant presence of hydroxyl groups in C5 and C7 sites of ring A implies changes in the molecular geometry, as both dihedral angles φ1 (−22.7°) and φ2 (134.1°) are wider than those found for the other compounds above discussed. The radicalization of compound 4 yields four radicals in positions C3′–OH, C4′–OH, C5–OH and C7–OH. Similarly to the other compounds, the C3′–OH radical results more stable than C4′–OH by less than 1 kcal mol−1, while the HAT reaction from both OH groups in the coumarinic portion of compound 4 leads to radical species significantly less stable than C3′–OH (6.12 and 7.50 kcal mol−1 for C5–OH and C7–OH, respectively).
The last of the previously synthesized compound (5) is characterized by a hydroxyl group in C8 and a bromine substituent in the C6 site of the coumarinic portion of the molecule. The presence of an electron withdrawing group in position C6 implies a more planar structure than those found for compounds 1–4, being both dihedral angles φ1 (−15.7°) and φ2 (123.3°), the smallest found. From compound 5 it is possible to obtain three radicals by HAT from C3′–OH, C4′–OH and C8–OH groups. The introduction of the above mentioned substituents, also in this case, does not influence the formation of the most stable radical found for the other compounds, as the radicalization of C3′–OH group remain the most likely one. In particular, the radicals' stability order is similar to what was found for compound 3, since the H-abstraction from C4′–OH and OH group on ring A (C6–OH and C8–OH for compound 3 and 5, respectively) leads to radicals less stable by about 0.1 and 5.0 kcal mol−1, respectively.
In all the cases examined so far, the free radical yielded by HAT from the C3′–OH position results more stable than that at C4′–OH, although in some cases by less than 1 kcal mol−1. This may be due to the hydrogen bond involving C3′–OH and C4′–OH which inhibits the H-abstraction from the C4′–OH group, and favours the C3′–OH radical. In addition, the electron withdrawing nature of the group in site 1′ of ring C makes the HAT more likely at the hydroxyl group in meta position, site 3′, than at that occupying the para position, site 4′.
Other five compounds have been designed, including several hydroxyl groups in different positions on the coumarinic portion of the hybrids. These compounds, have not been synthetized yet, and are investigated in order to predict their potential antioxidant activity, that (if enhanced with respect to compounds 1–5) might motivate experimental efforts for their production.
Compound 6 is characterized by the highest number of hydroxyl groups, at C5, C6, C7 and C8 positions of ring C. The slightly distorted structure of the catechol ring remains approximately the same as that found in the optimized geometries of the above discussed compounds, though both dihedral angles φ1 (−18.4°) and φ2 (127.8°) are slightly larger. Radicalization of compound 6, via HAT, may occur at all OH sites (C5, C6, C7, C8, C3′ and C4′). Contrarily to what was found for the compounds analysed until now, in this case HAT from the C3′–OH group does not lead to the most stable radical. Instead, the radical at C5–OH position is the lowest in energy, although both C6–OH and C8–OH radicals are less stable only by 1.05 and 0.69 kcal mol−1, respectively. The radicalization of C7–OH leads to the least stable radical of ring A (3.45 kcal mol−1). However, this radical is still lower in energy than those formed in ring C at C3′ and C4′ positions with energies higher by 7.97 and 7.85 kcal mol−1, respectively.
Compound 7 is derived from compound 6 by replacing the hydroxyl groups in positions C5 and C6 with H-atoms, while compound 8 was obtained by replacing the hydroxyl groups at C7 and C8 positions. In compound 7, the absence of hydroxyl groups in positions C5 and C6 does not introduce significant changes in the optimized structure. Similarly, compound 8 is characterized by both dihedral angles φ1 (−19.0°) and φ2 (129.8°) which differ from those found in compound 6 by less than 2 degrees. Four radicals can be obtained by HAT from compound 7, with the following stability order 8-OH > 4′-OH > 3′-OH > 7-OH. Comparing the radicals stability trend found for compound 7 with that of compound 6 it becomes clear that, although the free radical formed at C8–OH position is the most stable one, the instability of both catechol hydroxyl groups are small compared to the other possible coumarinic hydroxyl radicalization. The largest stability of radical C8–OH is essentially due to the weaker H-bonding interaction between such hydroxyl group and the heteroatom of the pyrone ring, which abstraction requires the least amount of energy.
HAT from the hydroxyl groups at C6, C5, C3′ and C4′ positions in compound 8 can produce four different radicals. The stability trend found in this case is C5–OH > C6–OH > C3′–OH > C4′–OH. Differently from compound 7, the radicals on the catechol part turn out to be the most unstable radicals, though they are less stable than the C6–OH one by about 4 kcal mol−1.
Compound 9 has been derived from compound 6 by substituting the OH at C7 with a methyl group, while in compound 10 both OH groups (at C6 and C7) are replaced by methyl ones. Thus, compounds 9 and 10 are characterized by three and two hydroxyl groups on ring A, respectively. HAT from compound 9 leads to five radicals, whose stability order was found to be C5–OH > C6–OH > C8–OH > C4′–OH > C3′–OH. The H-abstraction from 5-OH group results the most probable one, being its H atom the only one which is placed out of the coumarinic ring plane and that is not involved in hydrogen bonding. The presence of an electron donor group like methyl in C7 does not change the mutual position of coumarinic and catecholic rings being both dihedral angles φ1 (−18.1°) and φ2 (127.7°), i.e. essentially equal to those computed for compound 6, but its steric hindrance causes the opening of the C–C–O angles. In fact, all the four θ angles reported in Table 1 are wider in compound 9 than the equivalent ones in compound 6. Compound 10 produces four radicals HAT, corresponding to C5–OH, C8–OH, C3′–OH and C4′–OH groups. Also in this case, radicalization of 5-OH group leads to the most stable radical. The presence of two CH3 groups in positions C6 and C7 has an effect on the molecular geometry of the coumarin–chalcone derivative 10 similar to that observed in compound 9.
Similarly to the five compounds analysed above, radicalization in compounds 6–10 is favoured in those OH sites in which either its hydrogen is not directly interacting with neighbour oxygen atoms (compounds 6–8) or is in meta position with respect to the methyl group (compounds 9 and 10).
The spin densities of compounds 1–5, for which the most stable radical was demonstrated to involve the 3′-OH group of the catechol portion, show delocalization of the unpaired electron on the aromatic ring of the catechol moiety (see Fig. S3 of ESI†). However, when there are no hydroxyl groups on the coumarinic portion of the molecule, as in compounds 1 and 2, the C3′– and C4′–OH radicals result almost equally stable. On the other hand, the most favourable radicalization site for compounds 6–10 is different depending on the structure of the coumarin–chalcone derivative. However, for none of the designed derivatives the formation of the free radical is preferred on the catecholic portion of the molecule. Therefore, the computed spin density of all the most stable radicals show the same delocalization of the unpaired electron on the coumarinic portion of the molecules.
To summarize, when two or more adjacent hydroxyl groups are present on the coumarinic portion of such derivatives (compounds 6–9), the larger delocalization of the unpaired electron indicates that radicalization of the OH groups located on the coumarinic part are the most probable one, as well as when the OH groups are in adjacent positions to electron donating groups (compounds 9 and 10). On the contrary, the most probable radicalization site is C3′–OH when the number of hydroxyl groups in the coumarinic portion of such molecules is low (compounds 1, 2, 3), when they occupy non adjacent positions (compound 4), and also when an electron withdrawing group, such as bromine, is present (compound 5).
Mechanisms analysis
All the antioxidant activity parameters computed for the most stable species, including radicals and anions, of all the coumarin–chalcone derivatives are reported in Fig. 1, while those calculated for all the possible radicals and anions of each compound are collected in Table S4 of ESI.† In both Fig. 1 and Table S4† the antioxidant activity parameters computed for both DHM coumarin (6,7-dihydroxy-4-methyl-coumarin) and helichrysetin (4,4′,6′-trihydroxy-2′-methoxy-chalcone) were included for comparison.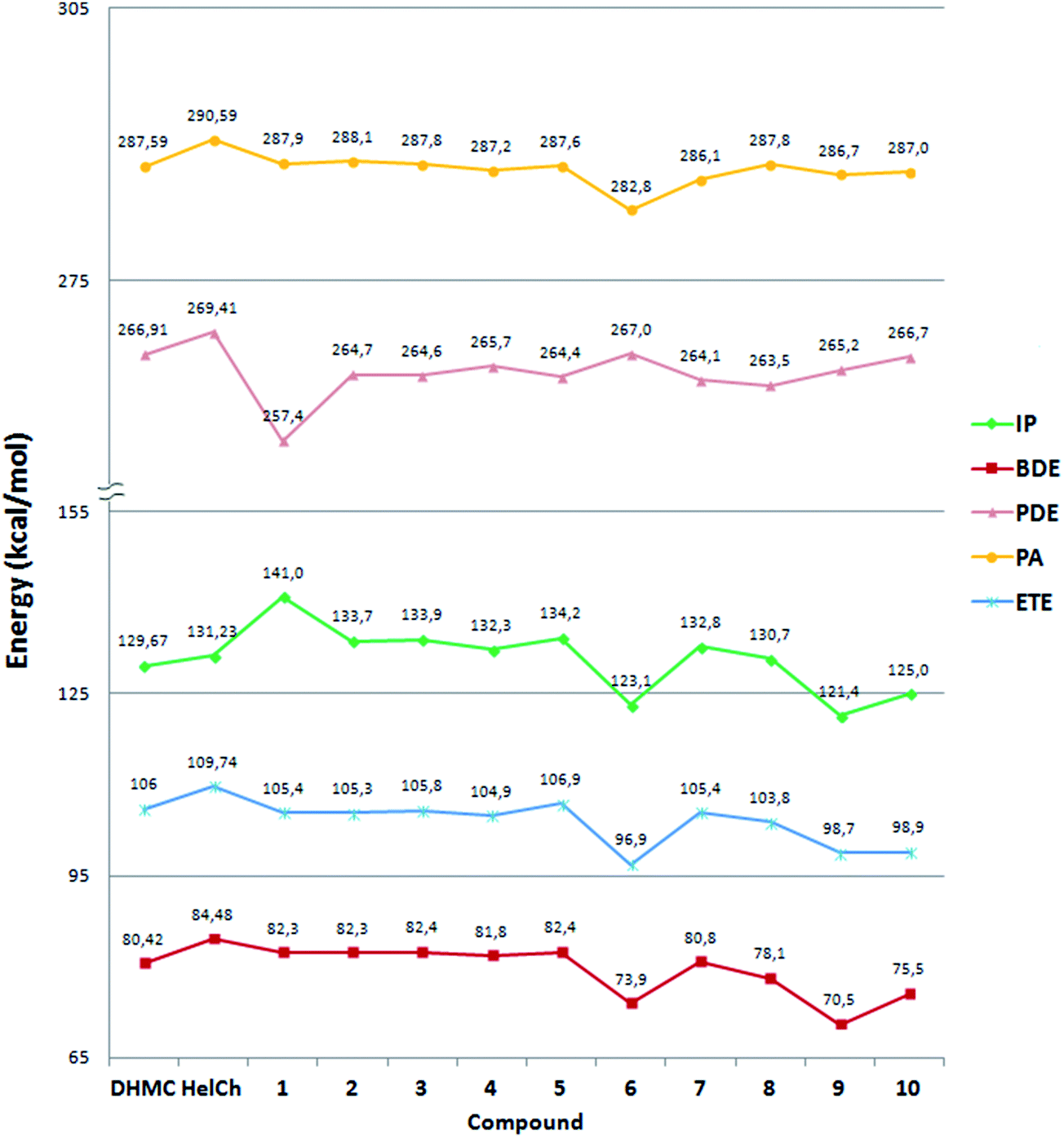 | ||
| Fig. 1 Thermodynamic parameters for the most stable species of each compound: bond dissociation enthalpies (BDE, red curve), electron transfer enthalpies (ETE, blue curve), adiabatic ionization potentials (IP, green curve), O–H proton dissociation enthalpies (PDE, pink curve) and proton affinities (PA, yellow curve) of the phenoxide anions (ArO−), at 298.15 K. | ||
Fig. 1 shows that the antioxidant parameters span in an energy scale ranging from 70 to 290 kcal mol−1, the lowest of which are the BDE values. Indeed, the BDE is the best reliable thermochemical parameter to describe the HAT mechanism, during which an H atom is transferred, from a hydroxyl group in the antioxidant compound, to the free radical. Therefore, the weakest O–H bond (lowest BDE) is expected to lead to the most likely reaction and then to the greatest antioxidant activity.
Fig. 1 shows that among the synthesized compounds, the labelled 4 coumarin–chalcone derivative seems to be the best antioxidant since it exhibits the lowest value of BDE, via HAT from the C3′–OH site. For all the five synthesized derivatives this dissociation process requires an amount of energy ranging from 81.76 kcal mol−1 (compound 4) to 83.97 kcal mol−1 (compound 2), which is a very small range. Therefore comparing the BDE values of derivatives 1, 2 and 3 shown in Fig. 1, it is possible to propose that while the presence of the methyl group in position C6 (compound 2) does not introduce any promoting effect on the antioxidant power with respect to compound 1, the introduction of an OH group in such position (compound 3) makes the O–H bond of the C3′–OH weaker than that present in the other molecules under comparison.
All the newly considered compounds (6–10) show a similar or even better antioxidant power than those already synthesized (1–5). In fact, the BDE values of such molecules range from 70.45 kcal mol−1 (compound 9) to 80.75 kcal mol−1 (compound 7), which are lower than the BDEs computed for compounds 1, 2, 3 and 5 and similar to that of compound 4. Therefore, compound 9, which exhibits the lowest BDE value, is proposed as a better antioxidant candidate, via HAT, than those already synthesized. This behaviour is clearly due to the concomitant presence of more than one hydroxyl group on the coumarinic portion of the designed molecules, distributed differently than in the previously reported compounds.
In addition, comparing the calculated data for the hybrid compounds with those corresponding to DHM coumarin and helichrysetin it becomes evident that all the studied hybrid species present BDE values that are lower than that of helichrysetin. This is a logical finding because the hybrids present a catechol moiety that promotes HT, due to the H bond interaction of the radicalized O atom with the H atom in the neighbour OH group, while helichrysetin does not present this structural feature. The presence of the OH in ortho position also increase reactivity because of its electron donor character. Regarding comparisons with DHM coumarin, compounds 1–5 have higher BDE values, while compounds 6–10 have similar (7) or lower (6, 8, 9 and 10) BDE values. On the contrary, according to the ORAC assay, hybrids 1 to 5 are better antioxidants than DHM coumarin and Helichrysetin, with compounds 4 and 5 being the best, and presenting similar activity based on the reported uncertainties. The best performance of these compounds, within the 1–5 set, is in agreement with the calculated data.
The apparent contradiction between ORAC and calculated data is due to the fact that while BDE values give important information regarding the intrinsic ability of a particular compound to donate an H atom, they cannot be directly compared with experimental values that arise from the whole reactivity of the analysed chemical species. There are several reasons for that. First of all the ORAC assays were carried out at pH = 7.4. Therefore due to the phenolic moieties present in the studied compounds a non-negligible fraction of them should be deprotonated, i.e., as mono-anions, which have a different (and probably higher) reactivity towards free radicals. In addition all the studied compounds have more than one phenolic site, thus they may react through different channels of reactions, with different contributions to the overall reactivity. Moreover, even though the ORAC assay is assumed to correspond to the HAT mechanism, contributions from reactions involving electron transfer cannot be ruled out. Probably a full kinetic study simultaneously including all the possible mechanisms and reaction sites would be useful to understand in more detail the observed behaviour.
The stepwise mechanisms, SET-PT and SPLET, are both related to the propensity of molecules to donate electrons. In the former mechanism, electron transfer leads to the formation of a radical cation ArOH+˙, which subsequently deprotonates. Therefore, ionization potential (IP) and proton dissociation enthalpy (PDE) are useful to establish trends regarding the viability such a mechanism. In the SPLET mechanism, on the other hand, a proton transfer takes place before the electron transfer, thus proton affinity (PA) and electron transfer enthalpy (ETE) are the most relevant properties for analysing the feasibility of this mechanism.
Analysing the IP and PDE values reported in Fig. 1, the energy amount required to form the radical cation is significantly lower than that necessary to accomplish the SET-PT second step. Thus, the last one seems to be the limiting step of such mechanism. On the contrary, the formation of the phenoxide anion during the first step of the SPLET mechanism (see PA values in Fig. 1) requires much more energy to occur than the electron transfer from the phenoxide anion to the free radical, indicating that for this mechanism the first step should be the slowest one.
Understanding why a mechanism is favoured over another ultimately requires the exploration of kinetics. However, that escapes the main purposes of the present work which intends to propose trends in reactivity for a relative large number of compounds of rather large size, considering several reaction sites and mechanisms. In addition, since the performed comparisons involve a series of closely related chemical processes, it seems justified to expect that the Bell–Evans–Polanyi principle is accomplished. In other words, at least when comparing reactions that take place through the same mechanism, a relationship between the activation energy and enthalpy of reaction is expected, and reactivity trends can be proposed based on thermochemical data.
Therefore, according to the data reported in Fig. 1 and Table S4,† it can be proposed that both stepwise mechanisms are less favourable than the HAT one, since the activation energies for both SET-PT (see PDE values) and SPLET (see PA values) mechanisms are expected to be higher than those of the HAT mechanism (described by BDE values). Therefore, the most likely mechanism by which coumarin–chalcone derivatives are proposed to exert their antioxidant activity, as inhibitors of the oxidation processes caused by free radicals, is the HAT mechanism. This is in good agreement with the results reported for other phenolic compounds.27,30,31 The other estimated parameters, associated to the SET-PT and SPLET mechanisms, could be useful in special conditions under which the HAT mechanism is unable to take place.
Other important information on the action mechanism of the investigated antioxidants can arise from the frontier orbital energies, EHOMO and ELUMO. The eigenvalues of LUMO and HOMO and their energy gaps, which reflect the chemical reactivity of the studied molecules, are reported in Table S5 of ESI.† The LUMO eigenvalue is directly associated with the electron acceptor ability (the lower the LUMO energies, the higher the electron accepting capacity), while the HOMO eigenvalue is used to evaluate the electron donor ability of molecular systems (the higher the HOMO energies, the better the electron donating capability). In addition, the smaller the HOMO–LUMO energy gaps, the easier the excitation of the HOMO electrons. The HOMO–LUMO gap for compound 9 was found to be the smallest one. Consequently, the electrons promotion from HOMO to LUMO in compound 9 is relatively easier to occur than in the other investigated derivatives. Data reported in Table S5† are, thus, in agreement with the IP adiabatic trend, confirming that compounds 9 is the best electron donor among the studied compounds. Therefore, hypothetically, it could work well following the SET-PT mechanism in which the removal of the hydrogen atom occurs only after one electron is transferred.
The HOMOs and LUMOs plots reported in Fig. S6 of ESI† show the typical π-like molecular orbital characteristics. While in compounds 1–5 and 7 the plot of HOMOs show a localization on the catecholic portion of the molecule, in compounds 6 and 8–10 the HOMO is localized on the coumarinic portion of such derivatives, anyhow in both cases an involvement of the carbonyl group emerges. In contrast, the LUMO is outspreaded on the coumarinic portion with small contributions on the substituents in all the investigated compounds.
Since antioxidants can act as reducting agents, their antioxidant capacity can be predicted also on the basis of their oxidation potentials. Lower oxidation potential points to a higher antioxidant activity.35 The first oxidation potential values of all the coumarin–chalcone derivatives previously synthesized (compounds 1–5) were experimentally measured by Vazquez-Rodriguez and collaborators, using cyclic voltammetry.20 They are reported in Table 2, together with those computed in the present study.
The experimental data indicate compound 5 as the best antioxidant since it exhibits the lowest value of Epa, while the theoretical values computed for the first oxidation potentials show only very small variations. Therefore, in contrast to the experimental data which show appreciable energy differences for 1–5 compounds, making easy to establish a trend in their antioxidant power, it turns out to be very difficult to select the derivative with higher antioxidant power based on the Epa theoretical values. However, the calculated data suggest that among the synthesized compounds, hybrid 4 seems to be a better antioxidant than the other compounds. On the contrary, for the newly considered group, the differences in Epa span allowing to clearly identify compound 9 as the best antioxidant followed by compounds 10 and 6.
In this case it seems also important to call attention to the fact that the experiments for determining the oxidation potentials were carried out at pH = 7.4. The influence of this experimental condition is not included in the calculations, which may lead to significant differences between the calculated and experimental data. As above mentioned, it is possible that a fraction of the studied compounds can be deprotonate to some extension at pH = 7.4. This fraction would be different for each compound depending on their particular pKa values, while the oxidation potentials of the anionic species are expected to be lower than those of the neutral ones. Therefore, depending on the pKa of each compound, and on the differences in electron transfer ability of their anions, the trends in oxidation potentials can be significantly modified. On the other hand, the calculated data only includes the neutral species. The data obtained this way provide information on the intrinsic reactivity, via electron transfer, for the studied compounds. However to obtain a better agreement with the experimental data a much more complex analysis is necessary, which escapes the purposes of the present work.
For systems as complex as those studied in this work, reactivity indexes such as BDE, IP, PDE, PA and ETE are very important to assess trends in the intrinsic reactivity of isolated molecules, in a particular acid–base form. This is the first logical step when analysing chemical reactivity of a series of molecules of relative large sizes, because it allows identifying the most promising ones for a particular purpose. In this case the analyses based in these properties led to identify compounds as the best candidates for acting as antioxidants. Further studies are still necessary to address the whole observable behaviour of the chalcone–coumarine hybrids, regarding their free radical scavenging activity. Since such studies are rather complex and laborious, the information provided in the present work is expected to assist in a well-documented reduction of the number of calculations needed for such a task. In other words, based on our results, such studies can now be focused only on those compounds identified as the best electron and H donors.
UV-Vis spectra
The UV-Vis spectra of coumarin36,37 and chalcone38,39 derivatives have been studied with both experimental and theoretical approaches. On the contrary, only few studies on coumarin–chalcone hybrids are available.19 Due to the lack of experimental UV-Vis spectra for the compounds included in this study, we have performed a preliminary study with different exchange-correlation functionals (XC) to test their reliability (B3LYP, PBE0, and wB97XD) in predicting UV-Vis data. To this purpose we have used, as reference, the experimental data on the two units which constitute the considered hybrids, coumarin and chalcone. The obtained results are reported in Table 3. All the employed XC give absorption maxima close to the experimental counterparts, with mean unsigned errors equal to 26, 24, and 21 nm for B3LYP, PBE0, and wB97XD, respectively. Therefore, they all are suitable for predicting the UV-Vis data of this kind of compounds.| Compound | MO contribution | B3LYP | PBE0 | wB97XD | Exp | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔE | f | ΔE | f | ΔE | f | ||||||
| eV | nm | eV | nm | eV | nm | nm | |||||
| Coumarin | H → L (64%) | 4.10 | 303 | 0.181 | 4.20 | 295 | 0.212 | 4.38 | 283 | 0.302 | 310 |
| H−1 → L (62%) | 4.42 | 281 | 0.236 | 4.55 | 273 | 0.224 | 4.84 | 256 | 0.183 | 285 | |
| Chalcone | H → L (69%) | 3.62 | 343 | 0.861 | 3.73 | 332 | 0.888 | 4.04 | 307 | 0.968 | 312 |
| H−1 → L (52%) | 4.26 | 291 | 0.094 | 4.41 | 281 | 0.108 | 4.94 | 251 | 0.191 | 230 | |
| 4 | H → L (70%) | 3.31 | 375 | 0.166 | 3.48 | 356 | 0.350 | ||||
| H−1 → L (65%) | 3.42 | 362 | 0.370 | 3.52 | 352 | 0.232 | 3.77 | 329 | 0.532 | ||
| H−4 → L (50%); H−2 → L (36%) | 3.82 | 324 | 0.186 | 3.93 | 316 | 0.173 | 3.99 | 311 | 0.300 | ||
| H → L+1 (56%) | 4.20 | 296 | 0.102 | 4.37 | 284 | 0.113 | 5.53 | 224 | 0.111 | ||
| 9 | H → L (70%) | 2.90 | 427 | 0.019 | 3.02 | 410 | 0.022 | 3.50 | 354 | 0.034 | |
| H−2 → L (63%) | 3.44 | 360 | 0.362 | 3.56 | 349 | 0.354 | 3.84 | 323 | 0.326 | ||
| H → L+1 (52%); H−1 → L+1 (15%) | 3.81 | 325 | 0.207 | 4.02 | 308 | 0.121 | |||||
| H−1 → L+1 (62%) | 4.10 | 302 | 0.123 | 4.26 | 291 | 0.133 | 4.02 | 308 | 0.495 | ||
The Soret band of both coumarins and chalcones is mainly characterized by the transition HOMO → LUMO with contributions of the HOMO−1 → LUMO. On the one hand, the π-like HOMO−1, HOMO and LUMO plots of coumarin molecules, reported in Fig. S8 of the ESI,† show electron delocalization distributed on the two condensed rings (A and B). On the other hand, in the chalcone molecule the delocalization of the HOMO−1, HOMO and LUMO orbitals is of a different type. While the former shows a delocalization concentrated on the phenylpropenonic unit, in the latter the conjugation chiefly involves the phenylpropenal portion. On the contrary, the conjugation in the LUMO includes both rings (see Fig. S7 of ESI†).
In the coumarin–chalcone hybrids the behaviour of both fragments, coumarin and chalcone, has to be taken into account in the interpretation of the absorption spectra. The main photo-physical characteristics of the considered coumarin–chalcone derivatives, computed in methanol solvent, are summarized in Table 3 and Table S8 of the ESI,† while the corresponding molecular orbital plots are shown in Fig. S6.† In general, the absorption spectra of all the considered compounds show a broad band in the region between 300 and 450 nm generated by different electronic transitions. For all the investigated compound, the HOMO–LUMO transition, corresponding to a π–π* transition, has been systematically found at higher wavelengths, compared to both the corresponding coumarin and chalcone fragments. While in compounds 1–5 and 7 the HOMO is localized on the catecholic portion of the molecule, compounds 6, 8–10 are characterized by a localization on the condensed rings of the coumarinic portion. Contrariwise, in all cases the LUMO is distributed on the entire molecule.
Considering as a reference the structurally simplest of the studied compound (1), in which no substituents are present on ring A, it is noteworthy that the addition of a weakly electron donor group, such as –CH3 (2) or –OH (3), in position C6 (Scheme 1) does not significantly shift the absorption band. The same behaviour is observed when two –OH groups are added in C5 and C7 positions in order to obtain compound 4. In compound 5 the introduction of an electron donor group –OH at position C8 and an electron withdrawing group –Br at C6 contributes to red shift the transition. With the exception of 7 all the other newly systems (6, 8–10) that contain several substituents show significant red shifts.
In order to get further insights in the change of the electron density upon electronic transition, the density difference between the first excited state and the ground state have been computed and reported in Fig. 2.
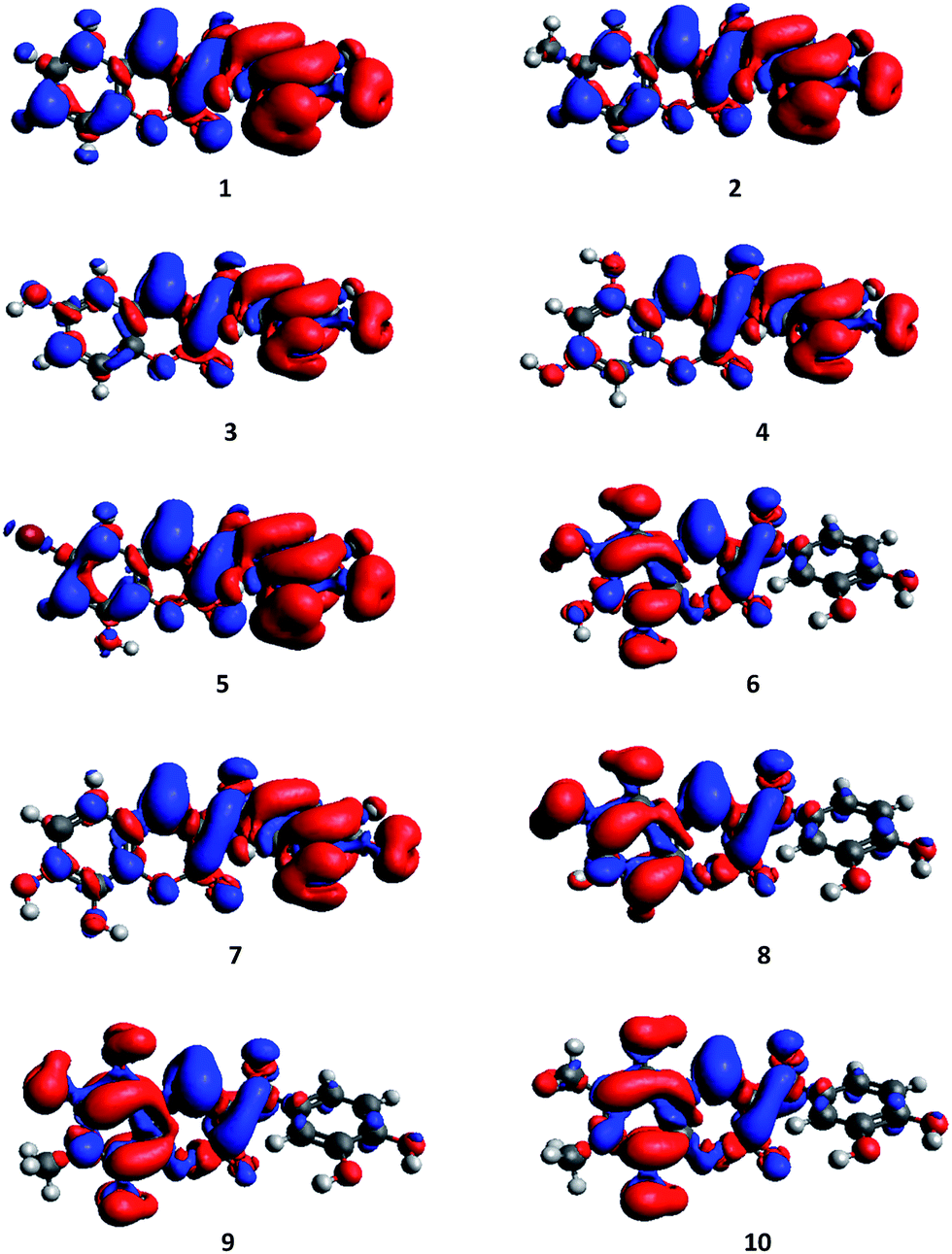 | ||
| Fig. 2 Computed density difference plots for coumarin–chalcone hybrids 1–10, with the first excited state considered (isodensity value of 1 × 10−3 a.u.); the blue color is associated with increase in electronic density upon electronic transition, while the red color is associated with decrease. | ||
In compounds 1–5 and 7 an increase of the electron density (blue region) upon the S0 → S1 transition appears in both phenyl and pyrone rings of the coumarins moiety, while a decrease (red color) is found in the other molecules. This means that a charge transfer from ring C to the coumarinic moiety takes place in the former compounds. On the other hand, in compounds 6 and 8–10 a charge transfer occurs from ring A to the pyrone ring B during the excitation as evidenced by the increase of the electron density on ring B and a decrease on ring A of the hybrids coumarinic portion.
Conclusions
Density functional based methods have been applied to study the antioxidant ability of a series of coumarin–chalcone derivatives. The main mechanisms proposed in the literature for the antioxidant action of polyphenols as radical scavengers were discussed in details, and structural and electronic features of species arising from these mechanisms were provided. The evaluation of the reaction enthalpies in condensed phase has been used as primary indicators of the radical scavenging activity exhibited by these compounds.The investigated coumarin–chalcone derivatives are not completely planar systems since in all cases ring C is twisted with respect to the rest of the molecule by around 20°. However, conjugation and delocalization of the π electrons occur in the two planar portions of the considered compounds. Among the investigated mechanisms, the HAT pathway is proposed as the most favourable, since it requires the lowest amount of energy to take place. All the considered compounds yield stable radical species upon the removal of a hydrogen atom in which the odd electron appears to be delocalized as much as possible on the portion of the molecule that is radicalized. The stability of such radicals is enhanced by the possibility that they establish internal H-bonds between the radicalized oxygen atom and vicinal hydroxyl groups, and by the electronic properties of the other groups in the conjugated system.
BDE values computed for the synthesized derivatives range from 81.76 to 83.97 kcal mol−1 in methanol, while the newly designed compounds exhibit BDE values ranging from 70.45 to 80.75 kcal mol−1, resulting better candidates to act as antioxidants than those already synthesized, especially when methyl substituents occupy adjacent positions to hydroxyl groups.
The evaluation of both the frontier orbital energies and the oxidation potentials confirm the IP adiabatic trend, according to which compound 9 is expected to be the best electron donor.
The simulated UV-Vis spectra of both coumarin and chalcone are in good agreement with the experimental counterpart, supporting the reliability of the spectra computed for the coumarin–chalcone hybrids investigated in this work, which show a broad band in the region between 300 and 450 nm generated by different electronic transitions.
Our investigation confirms the antioxidant properties of the recently synthesized coumarin–chalcone hybrids (1–5), and shows that poly-substitution of ring A enhances the antioxidant power of this class of compounds. Accordingly, our calculations strongly encourage the synthesis of such hybrids as an important strategy to develop novel compounds with improved antioxidant properties.
Acknowledgements
The University of Calabria and PON R&C (Programma Operativo Nazionale Ricerca e Competitività 2007–2013) project PON01_00293 “Spread Bio Oil” are gratefully acknowledged.Notes and references
- D. Batovska, S. Parushev, B. Stamboliyska and I. Tsvetkova, Eur. J. Med. Chem., 2009, 44, 2211–2218 CrossRef CAS PubMed.
- Y. L. Hsu, P. L. Kuo, W. S. Tzeng and C. C. Lin, Food Chem. Toxicol., 2006, 44, 704–713 CrossRef CAS PubMed.
- K. L. Lahtchev, D. I. Batovska, S. P. Parushev, V. M. Ubiyvovk and A. A. Sibirny, Eur. J. Med. Chem., 2008, 43, 2220–2228 CrossRef CAS PubMed.
- Z. Nowakowska, Eur. J. Med. Chem., 2007, 42, 125–137 CrossRef CAS PubMed.
- C. B. Patil, S. K. Mahajan and S. A. Katti, J. Pharm. Sci. Res., 2009, 1, 11–22 CAS.
- B. Orlikova, M. Schnekenburger, M. Zloh, F. Golais, M. Diederich and D. Tasdemir, Oncol. Rep., 2012, 28, 797–805 CAS.
- G. B. Bubols, D. R. Vianna, A. Medina-Remon, G. von Poser, R. M. Lamuela-Raventos, V. L. Eifler-Lima and S. C. Garcia, Mini-Rev. Med. Chem., 2013, 13, 318–334 CAS.
- R. J. Anto, K. Sukumaran, G. Kuttan, M. N. A. Rao, V. Subbaraju and R. Kuttan, Cancer Lett., 1995, 97, 33–37 CrossRef CAS.
- L. A. Pham-Huy, H. He and C. Pham-Huy, Int. J. Biomed. Sci., 2008, 4, 89–96 CAS.
- R. Torres, F. Faini, B. Modak, F. Urbina, C. Labbé and J. Guerrero, Phytochemistry, 2006, 67, 984–987 CrossRef CAS PubMed.
- K. N. Venugopala, V. Rashmi and B. Odhav, BioMed Res. Int., 2013, 1–14 CrossRef PubMed.
- B. Halliwell, Free Radicals and Other Reactive Species in Disease, in Encyclopedia of Life Sciences, 2001, pp. 1–7 Search PubMed.
- V. Lobo, A. Patil, A. Phatak and N. Chandra, Pharmacogn. Rev., 2010, 4, 118–126 CrossRef CAS PubMed.
- T. M. Florence, Aust. J. Ophthalmol., 1995, 23, 3–7 CrossRef CAS PubMed.
- P. G. Pietta, J. Nat. Prod., 2000, 63, 1035–1042 CrossRef CAS PubMed.
- F. Pérez-Cruz, S. Vazquez-Rodriguez, M. J. Matos, A. Herrera-Morales, F. A Villamena, A. Das, B. Gopalakrishnan, C. Olea-Azar, L. Santana and E. Uriarte, J. Med. Chem., 2013, 56, 6136–6145 CrossRef PubMed.
- K. V. Sashidhara, A. Kumar, M. Kumar, J. Sarkar and S. Sinha, Bioorg. Med. Chem. Lett., 2010, 20, 7205–7211 CrossRef CAS PubMed.
- K. V. Sashidhara, G. R. Palnati, R. Sonkar, S. R. Avula, C. Awasthi and G. Bhatia, Eur. J. Med. Chem., 2013, 64, 422–431 CrossRef CAS PubMed.
- Y. Xue, L. An, Y. Zheng, L. Zhang, X. Gong, Y. Qian and Y. Liu, Comput. Theor. Chem., 2012, 981, 90–99 CrossRef CAS PubMed.
- S. Vazquez-Rodriguez, R. Figueroa-Guíñez, M. J. Matos, L. Santana, E. Uriarte, M. Lapier, J. D. Maya and C. Olea-Azar, MedChemComm, 2013, 4, 993–1000 RSC.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A. 02, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- M. E. Alberto, N. Russo, A. Grand and A. Galano, Phys. Chem. Chem. Phys., 2013, 15, 4642–4650 RSC.
- M. Leopoldini, S. G. Chiodo, N. Russo and M. Toscano, J. Chem. Theory Comput., 2011, 7, 4218–4233 CrossRef CAS.
- M. Leopoldini, T. Marino, N. Russo and M. Toscano, J. Phys. Chem. A, 2004, 108, 4916–4922 CrossRef CAS.
- M. Leopoldini, T. Marino, N. Russo and M. Toscano, Theor. Chem. Acc., 2004, 111, 210–216 CrossRef CAS.
- M. Leopoldini, N. Russo and M. Toscano, J. Agric. Food Chem., 2006, 54, 3078–3085 CrossRef CAS PubMed.
- G. Mazzone, N. Malaj, N. Russo and M. Toscano, Food Chem., 2013, 141, 2017–2024 CrossRef CAS PubMed.
- G. Mazzone, M. Toscano and N. Russo, J. Agric. Food Chem., 2013, 61, 9650–9657 CAS.
- J. E. Bartmess, J. Phys. Chem., 1994, 98, 6420–6424 CrossRef CAS.
- E. Klein and V. Lukes, J. Phys. Chem. A, 2006, 110, 12312–12320 CrossRef CAS PubMed.
- M. E. Cassida, in Recent Advances in Density Functional Methods, Part I, ed. D. P. Chong, World Scientific, Singapore, 1995, p. 155 Search PubMed.
- R. Kohen and I. Gati, Toxicology, 2000, 148, 149–157 CrossRef CAS.
- R. H. Abu-Eittah and B. A. H. El-Tawil, Can. J. Chem., 1985, 63, 1173–1179 CrossRef CAS.
- J. Preat, D. Jacquemin and E. A. Perpète, Chem. Phys. Lett., 2005, 415, 20–24 CrossRef CAS PubMed.
- Y. Xue and X. Gong, J. Mol. Struct.: THEOCHEM, 2009, 901, 226–231 CrossRef CAS PubMed.
- H. H. Szmant and A. J. Basso, J. Am. Chem. Soc., 1952, 74, 4397–4400 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Born–Haber thermodynamic cycle used to calculate ΔG(sol); B3LYP optimized geometries of all the considered coumarin–chalcone hybrids; Spin densities of the most stable radical species formed by H removal from the neutral form of each compound; Bond dissociation enthalpies (BDE), O–H proton dissociation enthalpies (PDE), proton affinities (PA), electron transfer enthalpies (ETE) and adiabatic ionization potentials (IP) are reported in kcal mol−1; Energies of HOMO and LUMO for all compounds investigated, expressed in eV; Molecular surface contour plots for the highest occupied and lowest unoccupied molecular orbitals of all the investigated coumarin–chalcone derivatives; UV-Vis absorption data computed for coumarin and chalcone in methanol; Molecular surface contour plots for the two highest occupied and lowest unoccupied molecular orbitals of coumarin and chalcone. See DOI: 10.1039/c4ra11733f |
| This journal is © The Royal Society of Chemistry 2015 |