
Localized surface plasmon resonance enhanced quantum dot light-emitting diodes via quantum dot-capped gold nanoparticles†
Abstract
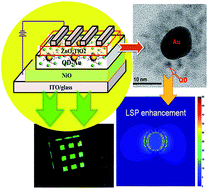
The coupling of localized surface plasmon resonance (LSPR) and excitons in nanomaterials can improve the performance of nano-optoelectronic devices. We synthesized a quantum dot (QD) capped gold (Au) nanoparticle (NP) composite to use as an emitting layer in all-inorganic quantum dot light-emitting diodes (QD-LEDs). The photoluminescence (PL) peak of the QDs is fully overlapped with the absorption peak of the Au NPs to realize the maximum LSPR enhancement. The LSPR effect of the QD-capped Au NPs has been demonstrated by increased radiative emission, PL excitation intensity, and reduced exciton lifetime evidenced by time-resolved PL measurement. As a result, a quantum yield of 60% for the QD-capped Au NPs has been realized and the QD-LED has achieved a remarkably enhanced power efficiency of 6.2 lm W−1 and maximum brightness of 1005 cd m−2 under various optimization conditions. These results demonstrate that QD-capped Au NPs constitute an effective route for achieving high-performance optoelectronic devices.
 Please wait while we load your content...
Please wait while we load your content...

