
Melamine based porous organic amide polymers for CO2 capture
Abstract
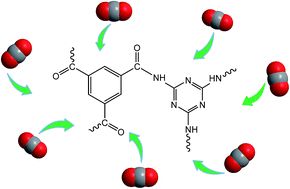
Amide based porous organic polymers were synthesized by the reaction of 1,3,5-benzenetricarbonyl trichloride with 2,4,6-triamino-1,3,5-triazine using two different solvents. Polyamide chains were derived from tri-functional monomers and their relative properties were compared in both media. These polymers were subjected to various analyses including FTIR, XRD, TGA, BET surface area and pore size analysis, FESEM and CO2 adsorption measurements. Thermal and chemical stability was achieved through strong amide building blocks in the polymer structure. The basic ring nitrogen and amide groups in the polyamide networks had the affinity to capture CO2. The maximum CO2 uptake of 2.99 cm3 g−1 (0.134 mmol g−1) at 273 K and 1 bar was obtained with the polyamide synthesized in DMAc–NMP (PA-1), revealing better efficiency than the polyamide prepared using 1,4-dioxane (PA-2) due to higher porosity and improved surface area. These thermally stable polyamides are anticipated to be good sorbents for CO2 capture in hostile environments.
 Please wait while we load your content...
Please wait while we load your content...

