
Uniaxial pressure induced phase transitions in multiferroic materials BiCoO3
Abstract
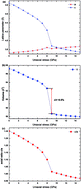
The crystallographic structure stability, spin state and electronic structure variation in tetragonal multiferroic material BiCoO3 under uniaxial pressure are investigated by means of first-principles density functional theory calculations. The lattice parameters, atomic internal coordinates and magnetic moment change abruptly under c axis compression of 9 GPa. A first-order structural phase transition occurs with a unit cell volume collapse of 9.5%, accompanied by the Co–O coordination polyhedron changing from CoO5 pyramid to the distorted CoO6 octahedron. A spin state transition of the Co3+ ions from the high-spin configuration in the CoO5 pyramidal coordination to the nonmagnetic low-spin configurations in the distorted CoO6 octahedron coordination has been explored. Contrasted electronic structure calculations are performed with PBE Generalized Gradient Approximation (GGA) and B3LYP hybrid functional. The hybrid functional drastically improves the band gap of the ground state. A controversial electronic structure has been predicted by GGA-PBE and B3LYP hybrid functional for the high pressure phase BiCoO3. We propose that the high pressure phase BiCoO3 is a nonmagnetic insulator.
 Please wait while we load your content...
Please wait while we load your content...

