
Living cationic ring-opening polymerization of 2-oxazolines initiated by rare-earth metal triflates†
Fangyu Hu,
Shoulei Xie,
Liming Jiang* and
Zhiquan Shen
MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: cejlm@zju.edu.cn
First published on 29th October 2014
Abstract
The cationic ring-opening polymerization (CROP) of substituted 2-oxazolines using rare-earth metal triflates (RE(OTf)3) as initiator was investigated for the first time. In this work, we examined the polymerization characteristics of 2-ethyl-2-oxazoline (EtOx) initiated by Sc(OTf)3 under conventional thermal heating and microwave irradiation, and compared the respective outcomes with those obtained with the most frequently used initiator methyl tosylate (MeOTs). The results indicated that Sc(OTf)3 exhibits a higher catalytic efficiency to the EtOx polymerization than MeOTs under identical conditions. The controlled/living nature of the Sc(OTf)3-catalyzed CROP was confirmed by its linear first-order kinetics and the narrow molecular weight distribution of the resultant polymers as well as the block copolymerization of EtOx and 2-phenyl-2-oxazoline (PhOx). Based on in situ NMR spectroscopic studies and SEC analysis of PEtOx samples obtained from the control termination experiments, a possible initiating/propagating mechanism has been proposed for the living cationic ring-opening polymerization. Morever, this rare-earth catalytic system can also be applied to the ring-opening polymerization of some sterically hindered or aryl-substituted 2-oxazolines.
Introduction
Poly(2-oxazoline)s are a class of synthetic polyamides that are receiving considerable attention in the past decade because of their synthetic versatility and potential applications in many fields, especially, in biomaterials.1–4 The polymerization of 2-oxazolines usually exhibits controlled and “living” character, i.e. it proceeds in the absence of chain transfer and termination reactions. This feature provides easy access to well-defined polymers with low polydispersity index (PDI) values and adjustable molar masses. Furthermore, a vast variety of substituted 2-oxazoline monomers, as well as functional initiators and terminating agents, is available and allows the tuning of the polymer properties.To date, various initiators including Lewis acids, such as boron trifluoride, and alkyl esters such as tosylates, triflates and halides have been reported for the cationic ring-opening polymerization of 2-oxazolines.5–7 Among them, tosylates and triflates could be particularly effective in combination with microwave irradiation, which enables the polymerization in minutes to a few hours.8,9 Although such a microwave-assisted polymerization was achieved in a laboratory equipment, the usefulness of this synthesis procedure and its manipulation still remain problematic for a scale up due to the intrinsic limitations of microwave techniques. As for tosylate and triflate, the yet most extensively used initiators, their high toxicity is a relevant key issue that must be taken in consideration in practical applications.10–13 Therefore, it is desirable to explore new catalytic systems to alleviate aforementioned problems and permit the CROP of 2-oxazolines to be performed on a normal apparatus. Also, we feel that more fundamental studies on the design and synthesis of polyoxazoline-based materials with new functional properties could be beneficial to the field.14–17
On the other hand, rare-earth metal triflates (RE(OTf)3) as a hard Lewis acid have been applied in the polymer synthesis. For example, Okamoto and his co-workers18–21 established a general method applicable to some polar monomers such as (meth)acrylamides and methacrylates wherein the added RE(OTf)3 catalytically affect the polymerization stereochemistry to isotactic-selective manner, producing corresponding highly stereoregular polymers. With the same protocol, we recently synthesized isotactic-rich polyacrylamide derivatives bearing a chiral oxazoline moiety in the side chain, which can serve as a chemosensor for the enantioselective recognition of 1,1′-bi-2-naphthol.22,23 More recently, RE(OTf)3 has proven to be an efficient catalyst for the ring-opening polymerization of lactones,24,25 tetrahydrofuran,26 and amino acid N-carboxyanhydrides.27 In view of this background, it was thought that this kind of rare-earth salts should have potential uses as initiator for the CROP of 2-oxazolines.
The present work aims at examining (i) the catalytic performance of rare-earth metal triflates for the cationic ring-opening polymerization of 2-oxazolines, and (ii) its monomer application scope. For this purpose, we investigated the Sc(OTf)3-catalyzed polymerizations of EtOx under the conventionally thermal heating and microwave irradiation conditions in comparison with those by using methyl tosylate as initiator. In addition, the influence of the temperature on polymerization kinetics was studied. Based on the observations from in situ NMR analyses and control termination experiments, a possible polymerization mechanism was proposed for the rare-earth catalysis. Finally, we briefly screened the polymerization behavior of other substituted 2-oxazolines in the presence of Sc(OTf)3. As depicted in Scheme 1, these monomers can be divided into two categories, namely, the sterically hindered 2-butyl-2-oxazolines bearing an alkyl group at the 4- or 5-position, and 2-phenyl-2-oxazoline derivatives where t-butyloxycarbonyl (Boc)-protected L-proline moiety was bound to the benzene ring via amide or ester linkage. Despite some Lewis acids had been utilized as an initiator for the polymerization of 2-oxazolines as early as 1960′s,28 we note that Lewis acidic rare-earth triflates have not yet been reported, to our knowledge.
 | ||
| Scheme 1 2-Oxazoline monomers tested in this study. | ||
Experimental section
Material
2-Ethyl-2-oxazoline (EtOx) and 2-phenyl-2-oxazoline (PhOx), purchased from Acros Organics, were dried and vacuum-distilled over barium oxide before use. Acetonitrile (CH3CN), dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF), methyl p-tosylate (MeOTs) and piperidine were distilled from calcium hydride. Piperazine (Aldrich) was distilled to use. Other chemicals, such as valeronitrile, 4-aminobenzonitrile, methyl salicylate, isatoic anhydride, dicyclohexylcarbodiimide (DCC), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), 4-dimethylaminopyridine (DMAP), N-Boc-L-proline, 2-aminobutanol, 2-aminopropanol, 1-amino-2-propanol, 2-chloroethylamine hydrochloride and ethanolamine (Aldrich) were used as received. Rare-earth metal triflates (RE(OTf)3, RE = Sc, Y, La, Dy, Lu) were prepared following the reported method29 and used after drying in vacuum.Analytical instrumentation and characterization techniques
1H NMR spectra were recorded on a Bruker Avance DMX-500 spectrometer. The chemical shifts are reported in ppm relative to tetramethylsilane (TMS) as an internal standard. Melting points were taken with a micro melting point apparatus (Shanghai Precision & Scientific Instrument Co. LTD, the accuracy = 0.1 °C). Size-exclusion chromatography (SEC) was measured on a Waters-150C apparatus equipped with two PLgel 5 μm MIXED-C 300 × 7.5 mm columns and a differential refractometer detector using DMF with LiBr (0.05 mol L−1) as the eluent (flow rate 1 mL min−1, 60 °C). The number-average molecular weight (Mn) and polydispersity index (PDI) of the polymers were calculated on the basis of a polymethylmethacrylate (PMMA) calibration. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra were acquired on a Bruker Ultraflextreme mass spectrometer in the positive reflector mode with an acceleration voltage of 25 kV, using sodium iodide (10 mg mL−1 in THF) as ionization salt and dithranol (20 mg mL−1 in THF) as matrix. Electrospray ionization-mass spectrometry (ESI-MS) measurements were performed with a Varian 500 mass spectrometer. The X-ray diffraction data for the complex Sc(OTf)3·EtOx were measured using an Atlas Gemini Ultima diffractometer (Mo-Kα radiation, ω-scan technique, λ = 0.71073 Å) at 170 K. The crystal structure was solved by direct methods using SHELXS-97.Monomer synthesis (Scheme 2)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), giving ProPhOx-1 (1.55 g, 86%) as a white crystal. M.p. = 123.2–124.2 °C. 1H NMR (400 MHz; CDCl3): δ = 1.47 (s, OC(CH3)3, 9H), 1.91–2.11 (m, N(CH2)2CH2, 2H), 2.23–2.59 (m, NCH2CH2CH2, 2H), 3.41–3.65 (m, NCH2(CH2)2, 2H), 4.02 (t, NCH2CH2O, 2H), 4.35 (t, NCH2CH2O, 2H), 4.51–4.60 (m, OCCHN, 1H), 7.08–7.19 (m, Ph, 1H), 7.27–7.32 (m, Ph, 1H), 7.45–7.52 (m, Ph, 1H), 7.91–7.95 (m, Ph, 1H); 13C NMR (125 MHz; DMSO-d6): δ = 22.9, 23.9, 28.0, 29.3, 46.3, 54.5, 58.8, 66.7, 79.0, 120.9, 123.3, 126.2, 130.3, 132.5, 148.9, 153.7, 160.2, 170.4. MS (ESI+): m/z (%) = 383.0 (39.6) [M + Na]+, 743.1 (100) [2M + Na]+.:1) as an eluant, and the intermediate 2a was obtained as a white crystal (1.84 g, 56.7%). M.p. = 53.4–54.3 °C (lit.31 54.5–55.5 °C). The amidation of 2a was conducted by the same procedure as the esterification of 1b, and the desired product ProPhOx-2 was obtained as a white crystal (89.5%). M.p. = 130.3–131.5 °C. 1H NMR (400 MHz; CDCl3), δ = 1.32 (s, OC(CH3)3, 9H), 1.91–1.95 (m, N(CH2)2CH2, 2H), 2.13–2.40 (m, NCH2CH2CH2, 2H), 3.40–3.72 (m, NCH2(CH2)2, 2H), 4.02–4.49 (m, NCH2CH2O, NCH2CH2O and OCCHN, 5H), 7.09–7.13 (m, Ph, 1H), 7.46–7.48 (m, Ph, 1H), 7.84–7.87 (m, Ph, 1H), 8.80–8.83 (m, Ph, 1H), 12.79 (s, NHCO, 1H); 13C NMR (125 MHz; DMSO-d6): δ = 23.2, 23.9, 27.7, 28.0, 30.2, 31.0, 46.4, 54.3, 61.9, 66.3, 78.8, 112.8, 118.5, 122.4, 128.9, 132.5, 139.0, 152.9, 153.9, 163.4, 172.1. MS (ESI+): m/z (%) = 382.1 (93.8) [M + Na]+, 741.1 (100) [2M + Na]+.:1) to give 3a as yellowish solid (3.67 g, 45.4%). M.p. = 159.5–160.5 °C (lit.32 160–161 °C). ProPhOx-3 was prepared by a similar way to that of ProPhOx-2 except for the use of EDC as the dehydrant and THF as solvent (53.7%, white crystal). 1H NMR (400 MHz; CDCl3): δ = 1.44 (s, OC(CH3)3, 9H), 1.84–1.98 (m, N(CH2)2CH2 and NCH2CH2CH2, 4H), 3.22–3.61 (m, NCH2(CH2)2, 2H), 4.04 (t, NCH2CH2O, 2H), 4.40–4.44 (m, NCH2CH2O and OCCHN, 3H), 7.56–7.59 (m, Ph, 2H), 7.88–7.90 (m, Ph, 2H), 9.77 (br, NHCO, 1H); 13C NMR (125 MHz; DMSO-d6): δ = 171.6, 162.6, 153.2, 141.7, 128.5, 122.1, 118.5, 78.5, 67.2, 60.2, 54.3, 46.6, 33.3, 30.9, 30.1, 27.9, 23.6. MS (ESI+): m/z (%) = 360.1 (100) [M + H]+, 719.0 (62.6) [2M + H]+, 741.1 (23.8) [2M + Na]+.
:1), giving ProPhOx-1 (1.55 g, 86%) as a white crystal. M.p. = 123.2–124.2 °C. 1H NMR (400 MHz; CDCl3): δ = 1.47 (s, OC(CH3)3, 9H), 1.91–2.11 (m, N(CH2)2CH2, 2H), 2.23–2.59 (m, NCH2CH2CH2, 2H), 3.41–3.65 (m, NCH2(CH2)2, 2H), 4.02 (t, NCH2CH2O, 2H), 4.35 (t, NCH2CH2O, 2H), 4.51–4.60 (m, OCCHN, 1H), 7.08–7.19 (m, Ph, 1H), 7.27–7.32 (m, Ph, 1H), 7.45–7.52 (m, Ph, 1H), 7.91–7.95 (m, Ph, 1H); 13C NMR (125 MHz; DMSO-d6): δ = 22.9, 23.9, 28.0, 29.3, 46.3, 54.5, 58.8, 66.7, 79.0, 120.9, 123.3, 126.2, 130.3, 132.5, 148.9, 153.7, 160.2, 170.4. MS (ESI+): m/z (%) = 383.0 (39.6) [M + Na]+, 743.1 (100) [2M + Na]+.:1) as an eluant, and the intermediate 2a was obtained as a white crystal (1.84 g, 56.7%). M.p. = 53.4–54.3 °C (lit.31 54.5–55.5 °C). The amidation of 2a was conducted by the same procedure as the esterification of 1b, and the desired product ProPhOx-2 was obtained as a white crystal (89.5%). M.p. = 130.3–131.5 °C. 1H NMR (400 MHz; CDCl3), δ = 1.32 (s, OC(CH3)3, 9H), 1.91–1.95 (m, N(CH2)2CH2, 2H), 2.13–2.40 (m, NCH2CH2CH2, 2H), 3.40–3.72 (m, NCH2(CH2)2, 2H), 4.02–4.49 (m, NCH2CH2O, NCH2CH2O and OCCHN, 5H), 7.09–7.13 (m, Ph, 1H), 7.46–7.48 (m, Ph, 1H), 7.84–7.87 (m, Ph, 1H), 8.80–8.83 (m, Ph, 1H), 12.79 (s, NHCO, 1H); 13C NMR (125 MHz; DMSO-d6): δ = 23.2, 23.9, 27.7, 28.0, 30.2, 31.0, 46.4, 54.3, 61.9, 66.3, 78.8, 112.8, 118.5, 122.4, 128.9, 132.5, 139.0, 152.9, 153.9, 163.4, 172.1. MS (ESI+): m/z (%) = 382.1 (93.8) [M + Na]+, 741.1 (100) [2M + Na]+.:1) to give 3a as yellowish solid (3.67 g, 45.4%). M.p. = 159.5–160.5 °C (lit.32 160–161 °C). ProPhOx-3 was prepared by a similar way to that of ProPhOx-2 except for the use of EDC as the dehydrant and THF as solvent (53.7%, white crystal). 1H NMR (400 MHz; CDCl3): δ = 1.44 (s, OC(CH3)3, 9H), 1.84–1.98 (m, N(CH2)2CH2 and NCH2CH2CH2, 4H), 3.22–3.61 (m, NCH2(CH2)2, 2H), 4.04 (t, NCH2CH2O, 2H), 4.40–4.44 (m, NCH2CH2O and OCCHN, 3H), 7.56–7.59 (m, Ph, 2H), 7.88–7.90 (m, Ph, 2H), 9.77 (br, NHCO, 1H); 13C NMR (125 MHz; DMSO-d6): δ = 171.6, 162.6, 153.2, 141.7, 128.5, 122.1, 118.5, 78.5, 67.2, 60.2, 54.3, 46.6, 33.3, 30.9, 30.1, 27.9, 23.6. MS (ESI+): m/z (%) = 360.1 (100) [M + H]+, 719.0 (62.6) [2M + H]+, 741.1 (23.8) [2M + Na]+.2-Butyl-2-oxazoline derivatives, including 4-ethyl-2-butyl-2-oxazoline (4-EtBuOx), 2-butyl-4-methyl-2-oxazoline (4-MeBuOx), and 2-butyl-5-methyl-2-oxazoline (5-MeBuOx) were synthesized according to a general procedure described in the literature.33
Polymerization procedure
All the polymerizations under thermal heating conditions were carried out on the Schlenk line with a predetermined initial monomer concentration. Taking the solution polymerization of EtOx as an example, a typical experimental procedure is as follows. To a flame dried flask EtOx (1 mL, 9.9 mmol) and 1.2 mL of an acetonitrile solution of Sc(OTf)3 (8.25 × 10−2 M) was added via a syringe under nitrogen atmosphere. The flask was then immersed in a constant-temperature oil bath of 80 °C. After being heated for a definite period of time with magnetic stirring, the reaction was quenched by the addition of deionized water. The resulting solution was left to stir at ambient temperature for 1 h and the crude product was precipitated in diethyl ether. To completely remove the catalyst residue and unreacted monomer, the collected powder matter was redissolved in CH3CN and then precipitated from Et2O. This process was repeated three times and the desired polymer PEtOx was dried under vacuum. The polymerization of other 2-oxazoline monomers and the purification are similar to that described above.For comparison, the polymerization of EtOx with microwave assistance was carried out in a CEM Discover SP microwave synthesizer. In this case, a stock solution of the monomer and Sc(OTf)3 or MeOTs as initiator with various monomer-to-initiator ratios was prepared in acetonitrile. The microwave vial was charged with 1–2 mL of this stock solution and heated in the microwave to a certain temperature. After a given time, the reaction mixture was cooled with compressed air and quenched by adding deionized water. The subsequent treatment of the polymerization product is the same as those described above.
Kinetic study of EtOx polymerization initiated with Sc(OTf)3
The kinetics of Sc(OTf)3-catalyzed cationic ring-opening polymerization of EtOx was studied under the conventionally thermal heating conditions in acetonitrile. In order to obtain the monomer conversion, aliquots taken periodically from the reaction mixture were directly subjected to 1H NMR analysis in CDCl3. The conversion was determined by comparing the signal intensity from the released CH2 of polymer at 3.6–3.3 ppm with that from the remaining CH2 of EtOx at 4.3 and 3.8 ppm (see: Fig. S1 in ESI†). Meanwhile, the collected samples were dissolved in DMF containing LiBr (50 mM) for SEC measurement to determine the molecular weight and polydispersity.Results and discussion
Monomer synthesis and polymerization features
Except for EtOx and PhOx, other 2-oxazoline derivatives shown in Scheme 1 were prepared from the commercially available starting materials (nitriles, carboxylates, or isatoic anhydride) by relatively facile functional-group transformations in moderate yields. Details of the synthetic preparations and structural characterization data of these monomers can be found in the experimental part and the ESI.†To evaluate the catalytic performance of rare-earth triflates for CROP of 2-oxazolines, we first investigated the solution polymerization of EtOx under different conditions. The polymerizations with RE(OTf)3 (RE = Sc, Y, La, Dy or Lu) were found to proceed smoothly at 80 °C in acetonitrile. As can be seen from Table 1 (runs 1–5), after a reaction period of 2 hours the monomer conversion of 47–84% was observed by 1H NMR analysis, yielding corresponding polymers with number-average molecular weight of 5270–8440 and low polydispersity indices (PDI = 1.09–1.12). Of five triflates tested, Sc(OTf)3 seems furnish best results in terms of the monomer conversion and PDIs of the resultant PEtOxs. In contrast, methyl tosylate (MeOTs) exhibited a poor initiating efficiency, which gave a monomer conversion of ∼16% and a low-molecular-weight polymer under the same conditions (run 6). Noteworthy is also the strong influence of the solvent property on the catalytic activity of Sc(OTf)3, with a decrease even deactivation observed when the polymerization was carried out in other solvents such as n-BuCl, DMSO or DMF instead of CH3CN (runs 7–9).
| Run | Catalyst | Solvent | [M]:[I] |
Temp. (°C) | Time (min) | PEtOx | ||
|---|---|---|---|---|---|---|---|---|
| Conv.c/% | Mnd | PDId | ||||||
| a The monomer concentration was 3 M for runs 1–9 and 4.5 M for runs 10–19.b The data given in parentheses are values obtained under microwave (μW) radiation.c Monomer conversion was determined by 1H NMR.d Determined by SEC, PMMA calibration, DMF containing 50 mM LiBr as the eluent.e Under μW conditions. | ||||||||
| 1 | Sc(OTf)3 | CH3CN | 100 | 80 | 120 | 84 | 5270 | 1.09 |
| 2 | Y(OTf)3 | CH3CN | 100 | 80 | 120 | 65 | 6220 | 1.12 |
| 3 | La(OTf)3 | CH3CN | 100 | 80 | 120 | 47 | 8440 | 1.12 |
| 4 | Dy(OTf)3 | CH3CN | 100 | 80 | 120 | 73 | 6270 | 1.10 |
| 5 | Lu(OTf)3 | CH3CN | 100 | 80 | 120 | 71 | 6290 | 1.11 |
| 6 | MeOTs | CH3CN | 100 | 80 | 120 | 16 | 3440 | 1.18 |
| 7 | Sc(OTf)3 | n-BuCl | 100 | 80 | 120 | 48 | 4230 | 1.21 |
| 8 | Sc(OTf)3 | DMSO | 100 | 80 | 120 | 0 | — | — |
| 9 | Sc(OTf)3 | DMF | 100 | 120 | 120 | 0 | — | — |
| 10b | Sc(OTf)3 | CH3CN | 100 | 80 | 60 | 53 | 4080 | 1.09 |
| (93) | (7880) | (1.10) | ||||||
| 11b | Sc(OTf)3 | CH3CN | 100 | 90 | 40 | 66 | 4490 | 1.10 |
| (91) | (8910) | (1.11) | ||||||
| 12 | Sc(OTf)3 | CH3CN | 100 | 140e | 5 | >99 | 5560 | 1.14 |
| 13 | MeOTs | 93 | 5320 | 1.27 | ||||
| 14 | Sc(OTf)3 | CH3CN | 400 | 140e | 20 | >99 | 12210 |
1.31 |
| 15 | MeOTs | 69 | 15320 |
1.52 | ||||
| 16 | Sc(OTf)3 | CH3CN | 800 | 140e | 20 | >99 | 19060 |
1.56 |
| 17 | MeOTs | 67 | 14850 |
1.97 | ||||
| 18 | Sc(OTf)3 | CH3CN | 1600 | 140e | 20 | 89 | 27240 |
1.67 |
| 19 | MeOTs | 44 | 14100 |
2.08 | ||||
In a set of comparable experiments, we found that the microwave (μW) irradiation makes Sc(OTf)3 more effective for the CROP of EtOx compared to the conventional heating, i.e., the rare-earth salt can catalyze the polymerization affording high yields (>90%) of higher-molecular-weight polymers with narrow PDIs under microwave conditions in acetonitrile at 80 or 90 °C (runs 10 and 11). Such a significant improvement in catalytic efficiency pointed to the existence of a non-heat microwave effect in the Sc(OTf)3-catalyzed polymerization. Nevertheless, for the reported μW-assisted CROPs of 2-oxazolines there seemed to be no distinct correlation between the accelerated polymerization rate and the microwave effect, as demonstrated by Hoogenboom and Schubert et al.34
It is also interesting to note that Sc(OTf)3 always exhibited higher catalytic efficiency than MeOTs in the μW-assisted polymerization process (runs 12–19). For example, the Sc(OTf)3-catalyzed reaction came to completion within 5–20 minutes at 140 °C in the cases of monomer-to-initiator ratio of 100, 400 and 800, even at a [M]/[I] of 1600 the monomer conversion reached ∼90% yielding Mn as high as 27000. However, with decreasing of the catalyst loading a relatively broad molecular weight distribution was observed for the resultant PEtOxs (PDI value changing from 1.14 to 1.67), which may be attributed to chain transfers during the CROP.35
Table 2 summarizes the bulk polymerization results of EtOx using Sc(OTf)3 as the initiator under conventionally thermal heating. It can be seen that this bulk polymerization process allowed the synthesis of polymers with molar masses in the range 5400 g mol−1 < Mn < 18800 g mol−1 (PMMA standard) through regulation of reaction parameters. In experimenting we found that with the proceeding of polymerization the reaction mixture became highly viscous and later solidified. It usually occurred at the point that the monomer conversion was 70–80%. Most likely, the presence of such a viscous or heterogeneous system at the late stage of polymerization is responsible for the broader molecular weight distributions (PDI = 1.27–1.37). This speculation is supported by the data from Table 2. More specifically, in the polymerization with a monomer-to-initiator ratio of 100 (runs 1–3), elongating the reaction time from 30 min to 60 min resulted in a distinct increase in both Mn and PDI; however, both the values almost remained constant when further extending periods of time. From the results shown in runs 6–8 of Table 2, it appears that the combination of a lower temperature and longer reaction time is beneficial to the improvement of molar masses while keeping narrower polydispersity for the case of high [M]/[I] ratio.
| Run | [M]:[I] |
Temp./°C | Time/min | PEtOx | ||
|---|---|---|---|---|---|---|
| Yielda/% | Mnb | PDIb | ||||
| a Isolated yield determined by gravimetry.b Determined by GPC, PMMA calibration, DMF containing 50 mM LiBr as the eluent. | ||||||
| 1 | 100 | 80 | 30 | 34.2 | 5450 | 1.09 |
| 2 | 100 | 80 | 60 | 89.0 | 12040 |
1.29 |
| 3 | 100 | 80 | 90 | 89.4 | 12920 |
1.29 |
| 4 | 100 | 100 | 30 | 86.4 | 8860 | 1.28 |
| 5 | 200 | 100 | 30 | 86.1 | 9030 | 1.37 |
| 6 | 500 | 100 | 30 | 70.1 | 13800 |
1.26 |
| 7 | 500 | 100 | 180 | 97.8 | 12410 |
1.64 |
| 8 | 500 | 80 | 180 | 85.0 | 18730 |
1.32 |
Kinetic studies of EtOx polymerization with Sc(OTf)3
The polymerization kinetics of EtOx were determined in acetonitrile with Sc(OTf)3 as the initiator under conventional conditions, using an initial monomer concentration of 4.5 M and a monomer-to-initiator ratio of 100. The resulting kinetic plots at five different polymerization temperatures are depicted in Fig. 1a. It can be seen that at 100 °C or 90 °C the monomer revealed linear first-order kinetics up to ∼90% conversion (ln[M]0/[M]t ≈ 2.3) in less than 2 h. For the polymerization conducted at 80 °C and 70 °C, this linear relationship was observable in the range of 86% and 68% conversion within 3 h, respectively. In all experiments, SEC analyses of the obtained polymers gave unimodal traces. As shown in Fig. 1b, the number-average molecular weight (Mn) increased linearly in proportion to the conversion, while the PDIs remained around 1.10. As such, it can be concluded that the polymerizations proceeded in a controlled/living manner. | ||
| Fig. 1 (a) Kinetic plots for 2-ethyl-2-oxazoline polymerizations initiated with Sc(OTf)3 ([M]0/[I]0 = 100) in acetonitrile ([M]0 = 4.5 M) at 60, 70, 80, 90, and 100 °C under thermal heating; the apparent rate constants (kp) are expressed in L mol−1 min−1. (b) Evolution of the molar mass (Mn) and the PDI value with monomer conversion (determined by SEC, RI detection, PMMA calibration, DMF containing 50 mM LiBr as the eluent. See: Fig. S2 in ESI†). | ||
The fact that ln[M]0/[M]t increased linearly with reaction time (Fig. 1a) indicates that the concentration of propagating species remained constant throughout the polymerization. Assuming that the concentration of the active species [P*] was equal to the initial initiator concentration [I]0, the formula (eqn (1)) for determining the apparent rate constant (kp) was integrated in eqn (2). Then, kp values can be deduced by the slope of the ln([M]0/[M]t versus time, as shown in Fig. 1a. From the graphical resolution of the Arrhenius equation (eqn (3) and Fig. 2), the activation energy (Ea) and the frequency factor (A) are calculated as 69.54 kJ mol−1 and 9.77 × 107 L mol−1 s−1, respectively. The Ea value is comparable to that obtained in the MeOTs initiating system.8
 | (1) |
 | (2) |
| kp = Ae−Ea/RT | (3) |
 | ||
| Fig. 2 Arrhenius plot for the polymerizations of 2-ethyl-2-oxazoline with Sc(OTf)3 in acetonitrile (see Fig. 1a for kp values). | ||
As evidenced by the kinetic results, Lewis acidic Sc(OTf)3 is more reactive towards EtOx polymerization than other existing initiators such as methyl tosylate8 and alkyl iodides.36 With the rare-earth catalyst the high yield synthesis of PEtOx was achieved in a rapid manner (within several hours) under mild conditions. Furthermore, this living system can be expected for the preparation of block copolymers with a well-defined structure. For instance, we have successfully synthesized a diblock polymer comprised of PEtOx and PPhOx segments via a sequential polymerization route, wherein the formation of copolymer was confirmed by the observed SEC analyses of a peak shift from 20.3 to 18.4 min elution time from the first (PEtOx) to second block growth profiles and a narrow molecular weight distribution (PDI = 1.18) (Fig. 3).
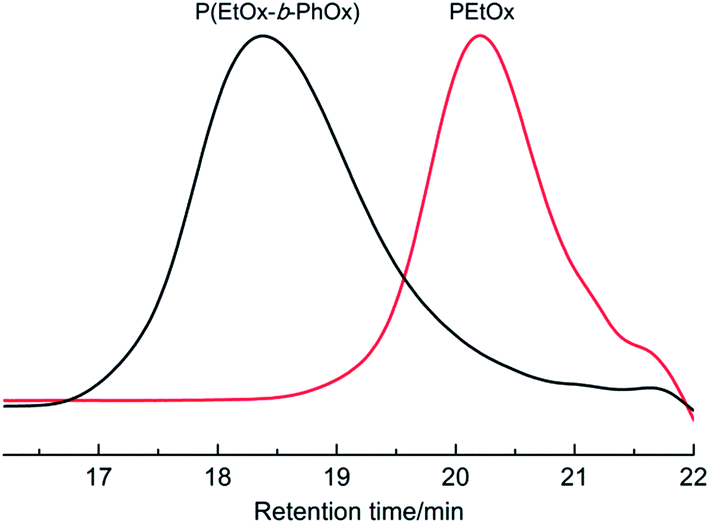 | ||
| Fig. 3 SEC profiles of PEtOx and the in situ prepared poly(EtOx-b-PhOx) by using the sequential addition method (SEC: RI detection, PMMA calibration, THF as eluent, with a flow rate of 1 mL min−1, 40 °C). Copolymerization: Sc(OTf)3–EtOx–PhOx = 1:28:60 (molar ratio), [M]total = 4.5 M, acetonitrile, 90 °C; the second monomer was added when EtOx conversion reached >99% (traced by 1H NMR). The Mn's of PEtOx and poly(EtOx-b-PhOx) were 1190 and 3340, respectively. | ||
Mechanistic aspects
The cationic ring-opening polymerization of 2-oxazolines proved to proceed via two different types of active species, i.e. ionic and covalent types, depending on the nature of the initiator and on the monomer.5 For the present system, the ionic polymerization mechanism seems to be most possible because the counter anion −OTf is of poor nucleophilicity. Morever, judging from the low polydispersity (PDIs ≤ 1.10) of the purified kinetic samples, the cationic species of an oxazolinium is likely to be the only propagating end existed during the polymerization. Further support to this speculation was given by analysis of the MALDI-TOF mass spectrum of PEtOx with piperidine terminals. These polymer samples were obtained by end-termination of the living oxazolinium species resulting from the CROP of EtOx with excess piperidine. As can be seen from Fig. 4, the mass difference in the series of signals (m/z 99.1) agrees well with the mass of one EtOx repeating unit, confirming that the sodium adducts form a homologue series originating from end-functionalized polymers. Similar results observations were made for the higher molecular weight polymers, shown in Fig. S3 in the ESI.† | ||
| Fig. 4 A typical MALDI-TOF mass spectrum of PEtOx having a piperidine group at the ω-terminal end (Mn,SEC = 3560, Mw,SEC/Mn,SEC = 1.11; PMMA standard, eluent: DMF with 50 mM LiBr). The inset shows the corresponding full spectrum. Polymerization conditions: [M]0 = 4.5 M, [M]0/[I]0 = 100, CH3CN, Sc(OTf)3 initiator, 90 °C, 1 h, piperidine terminator. | ||
Single-crystal X-ray diffraction analysis of Sc(OTf)3–EtOx complex revealed extensive coordinative interactions between the imine units of the monomer and the scandium cation (Fig. S4 in the ESI†). It is conceivable that the coordination would promote the nucleophilic attack of free monomers at the C-5 position of the bound oxazoline unit to generate an oxazolinium propagating species, which has been confirmed by in situ NMR spectroscopic studies. As shown in Fig. 5, the EtOx polymerization system before being subjected to the termination (i.e. a living polymer) displays the weak proton signals at 4.26 and 4.90 ppm (denoted as a and b). The two peaks should come from the active oxazolinium units, because they are different from those due to methylene protons of monomer (δ: 3.71, 4.17 ppm) and those of the polymer main chain (δ: 3.6–3.3 ppm) as well; furthermore, they vanished totally upon addition of terminating agents such as piperidine (see: Fig. S1 in the ESI†).
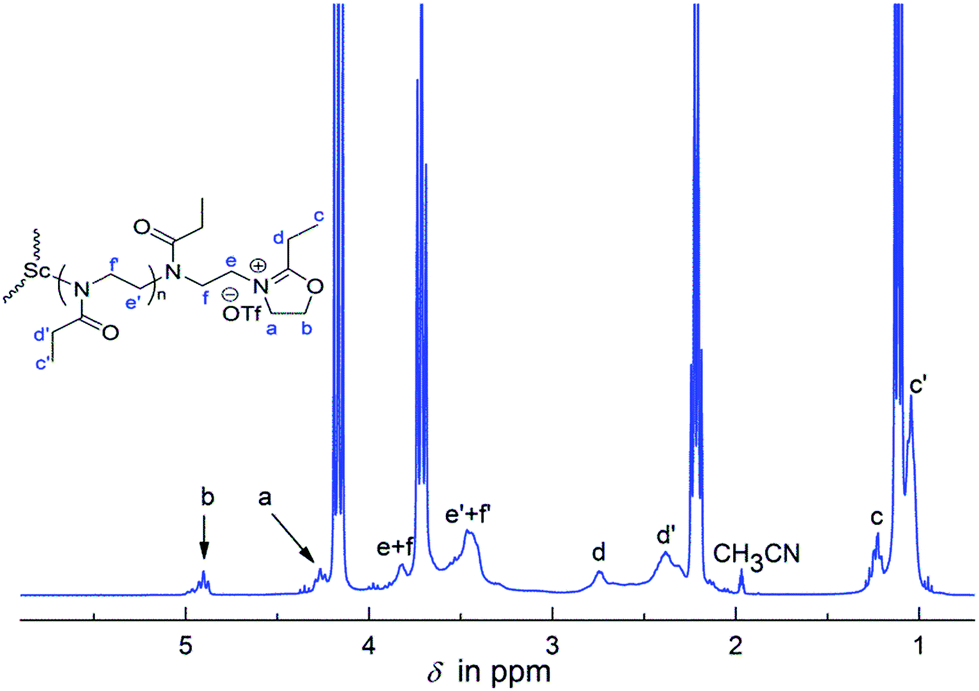 | ||
| Fig. 5 1H NMR spectrum of a living polymer sample formed in the EtOx polymerization in CD3CN at 80 °C for 30 min ([EtOx]/[Sc] = 30). | ||
Simultaneously, 13C NMR spectroscopy provides us with more information about the propagating species. In the 13C NMR spectra of Sc(OTf)3, the anionic ligand displays a characteristic quartet at 114.4–123.8 ppm, and the position of these signals does not change distinctly in the presence of EtOx at ambient temperature (Fig. 6a and b). However, a ca. 1.2 ppm downfield shift was observed upon heating the mixture at 80 °C for 30 min (Fig. 6c), indicating that the ligand moved to the propagating ends as a bound counterion with the polymerization went on. This situation is similar to that seen in the ligand exchange of rare-earth triflates reported by Ling et al.37
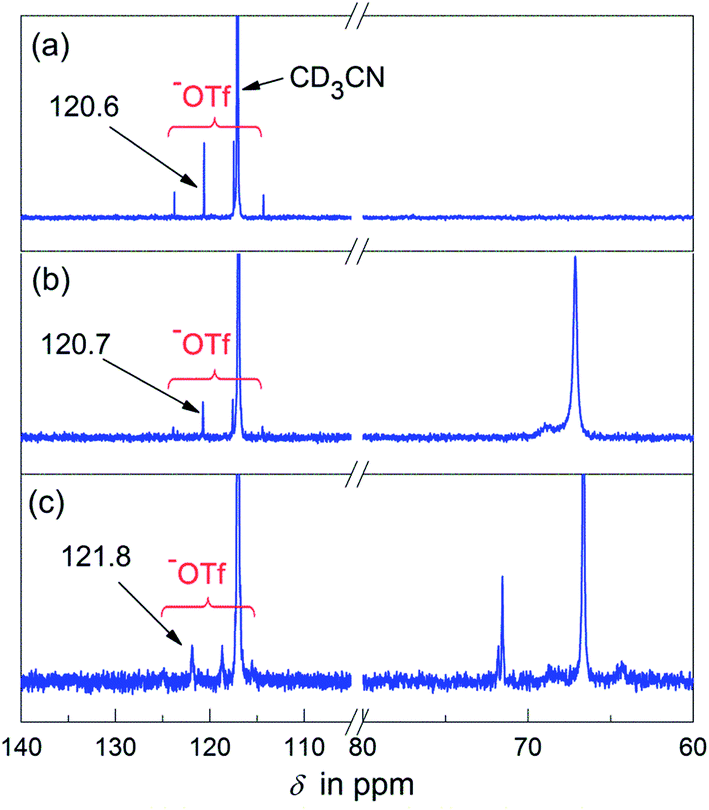 | ||
| Fig. 6 13C NMR spectra in CD3CN of (a) Sc(OTf)3, (b) Sc(OTf)3 + EtOx (30 eq.), 25 °C, and (c) the mixture composed of Sc(OTf)3 and EtOx (30 eq.) after heating for 30 min at 80 °C. | ||
It is noteworthy that the 13C NMR signal of the ligand −OTf still remained its intrinsic quartet feature in the living polymerization system (Fig. 6), which means they are in the identical chemical environments as counterions. Hence we presume that every metallic active species in situ-generated from the Sc(OTf)3–EtOx complexation may possess three identical chain-growing sites, in other words, one Sc(OTf)3 initiated three equivalent PEtOx chains. To clarify this point, we devised a set of control experiments, in which the parallel EtOx polymerizations initiated with Sc(OTf)3 under the same conditions ([M] = 4.5 M, [M]/[I] = 100, 90 °C, 60 min) were terminated by two kinds of terminating agents, the monofunctional piperidine and the bifunctional piperazine. Rather, the termination reaction was conducted at a predetermined terminator-to-initiator molar ratio ([T]/[Sc]) to prepare the desired polymers (P1–P5) for SEC analysis. Among them, P1, as a reference, was obtained by using intentionally excess piperidine ([T]/[Sc] = 6) in the termination step, while P2–P5 resulted from the piperazine termination with [T]/[Sc] of 0.5, 1.0, 1.5, and 2.0, respectively.
As shown in Fig. 7, P1 gives a unimodal molecular weight distribution with Mn = 3310 (PDI = 1.09) in THF. In contrast, the SEC traces of P2–P5 exhibit a bimodal shape and the peak of low molecular weight components (LMW, arbitrarily defined as Mn ≈ 3 kD) was found to decrease in going from P2 to P4 in concert with an increase in the terminator amount used in termination reactions. The Mn values of high molecular weight (HMW) polymers are roughly twice Mn's of LMW parts. These observations may be explained by the idea proposed above that one Sc(OTf)3 leads to three living polymer chains combination with Scheme 3 illustrating the initiating/propagating mechanism. In the piperazine-termination with [T]/[Sc] = 0.5, the nucleophilic sites (secondary amino groups) were not in sufficient quantities to stop all the living chains and thereby the resultant polymer P2 is composed of the piperazine-coupled HMW PEtOx and non-coupled LMW PEtOx, as evidenced by its SEC curve (Fig. 7). It was also the case for the termination with [T]/[Sc] = 1.0, but the relative content of LMW components in P3 decreased remarkably compared with P2.
 | ||
| Fig. 7 Normalized SEC traces (THF, RI detection) of PEtOx samples obtained by terminating the EtOx polymerization with piperidine (P1) and piperazine (P2–P5); the added amounts of terminators ([T]/[Sc]) are noted in the inset (PD = piperidine, PZ = piperazine). | ||
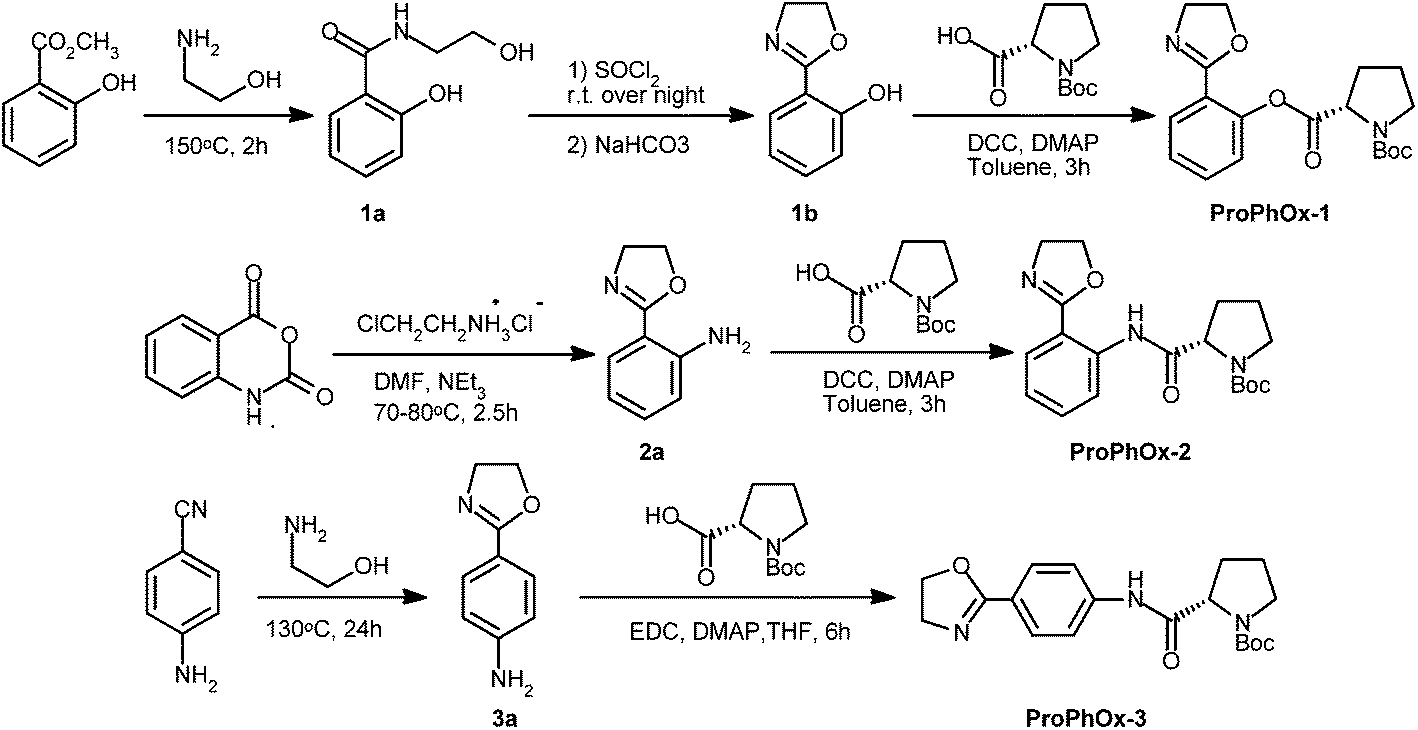 | ||
| Scheme 2 Synthesis of 2-phenyl-2-oxazoline (PhOx) derivatives bearing Boc-protected L-proline moiety. | ||
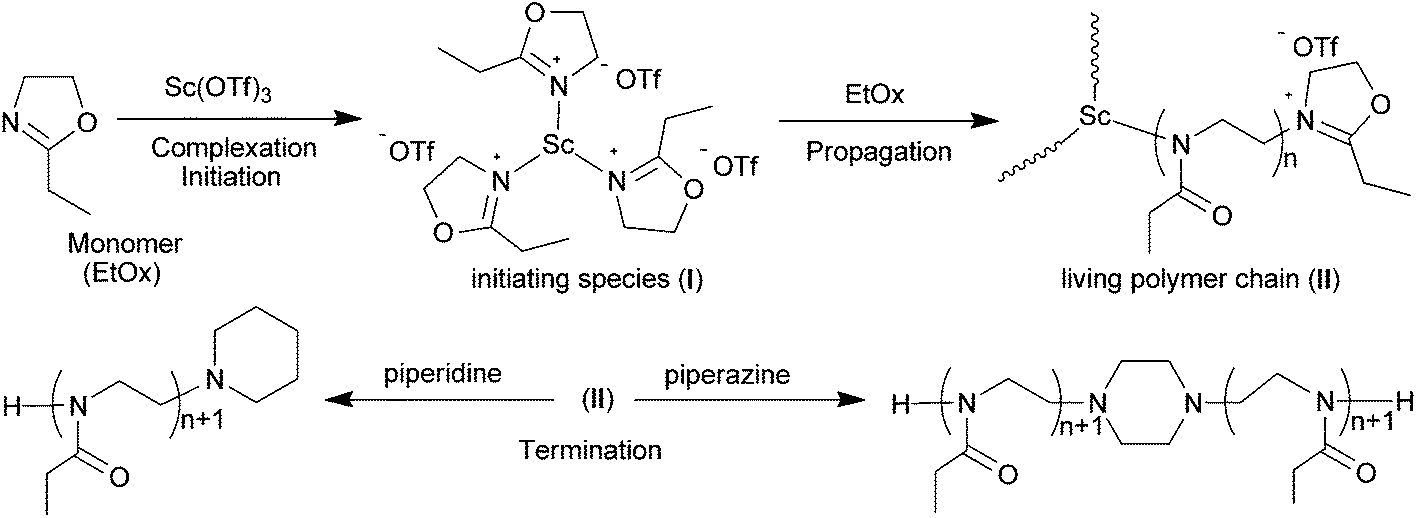 | ||
| Scheme 3 Schematic representation of the cationic ring-opening polymerization of 2-ethyl-2-oxazoline (EtOx) with Sc(OTf)3 as the initiator. | ||
Interestingly, the SEC elution trace of P4 is close to a unimodel shape, indicative of this sample consisted of the coupled polymers predominantly. This result seems to be understandable, because in the case of [T]/[Sc] = 1.5 the bifunctional terminator just provides equivalent nucleophilic cites relative to the cationic propagating ends, whereby every piperazine molecule can catch two active chains in average during the termination process yielding the coupled product. The very minor distribution occurred in the SEC profile reveals the presence of a few PEtOx polymers resulting from chain transfer to the moisture or/and nucleophilic impurity that enter reaction vessel. On the other hand, an excess of piperazine would retard such coupling reactions owing to partial terminators is bound to serve as the monofunctional capping agent in this case, which increasing the content of LMW polymers, as shown in SEC curve of P5 (Fig. 7).
Extension of Sc(OTf)3-catalysis to other substituted 2-oxazolines
Finally, we tried to extend the rare-earth catalysis to CROPs of some sterically hindered and functional 2-oxazoline derivatives. The preliminary results suggest that 4/5-alkylated 2-butyl-2-oxazolines (i.e., 4-EtBuOx, 4-MeBuOx and 5-MeBuOx) as well as 2-phenyl-2-oxazoline (PhOx) can be polymerized in a thermal heating process using Sc(OTf)3 as the initiator, leading to corresponding polymers with low molecular weights and a relatively narrower polydispersity (runs 1–4, Table 3). At a high catalyst loading ([M]/[I] = 20), the polymerization of ProPhOx-1 containing ester linkage was achieved at 90 °C, while its amide analogs (ProPhOx-2 and ProPhOx-3) did not give the desired polymers (runs 5–7). Different from ProPhOx-2, however, ProPhOx-3 can be polymerized at 140 °C under microwave irradiation resulting in the polymer with Mn of ∼6.0 kD and a narrower molecular weight distribution (PDI ∼ 1.4). The poor polymerizability of ProPhOx-2 is probably attributed to the fact that the intramolecular hydrogen bonding interaction between the adjacent oxazoline unit and amide group decreases the cyclic-imine basicity to such an extent that the oxazolinium initiating species failed to form. However, a relevant case was reported by Jordan and coworkers,13 who showed that methyl triflate did not efficiently initiate the ring-opening polymerization of a kind of 2-oxazoline monomer with a Boc protected amino function (2-[N-Boc-5-aminopentyl]-2-oxazoline). They presumed the initiator does not only attack the 2-oxazoline ring, but mainly attacks the secondary amide group in the monomer side chain.| Run | Monomer | [M] (mol L−1) | [M]:[I] |
Solvent | T/°C | t min−1 | Polymera | ||
|---|---|---|---|---|---|---|---|---|---|
| Yieldb/% | Mnc | PDIc | |||||||
| a The structural characterization of polymers are given in Supporting Information.b Isolated yield.c Determined by SEC, PMMA calibration, DMF containing 0.05 M LiBr as the eluent.d Under μW conditions. | |||||||||
| 1 | 4-EtBuOx | Neat | 60 | Bulk | 120 | 180 | 48 | 4650 | 1.13 |
| 2 | 4-MeBuOx | Neat | 60 | Bulk | 120 | 180 | 38 | 4530 | 1.15 |
| 3 | 5-MeBuOx | Neat | 60 | Bulk | 120 | 180 | 26 | 4230 | 1.20 |
| 4 | PhOx | 4.5 | 100 | CH3CN | 90 | 180 | 31 | 5510 | 1.21 |
| 5 | ProPhOx-1 | 1.0 | 20 | CH3CN | 90 | 180 | 85 | 2460 | 1.14 |
| 6 | ProPhOx-2 | 1.0 | 20 | CH3CN | 90 | 180 | — | — | — |
| 7 | ProPhOx-3 | 1.0 | 20 | CH3CN | 90 | 180 | — | — | — |
| 8 | ProPhOx-2 | 1.0 | 20 | CH3CN | 140d | 20 | — | — | — |
| 9 | ProPhOx-3 | 1.0 | 20 | CH3CN | 140d | 20 | 46 | 5840 | 1.42 |
Conclusions
In summary, we have demonstrated that rare-earth metal triflates could be used as an efficient catalyst for the cationic ring-opening polymerization of 2-oxazolines under mild conditions. As evidenced by linear first-order kinetics, the polymerization of 2-ethyl-2-oxazoline with Sc(OTf)3 in acetonitrile proceeded in a living/controlled manner, which was also supported by unimodel molar mass distribution for the resultant polymers with PDI values below 1.15. In situ NMR spectroscopic studies in combination with SEC analysis of PEtOx samples obtained from the control termination experiments showed strong evidence that during the Sc(OTf)3-catalyzed polymerization every in situ-formed metallic active species consists of three identical onium sites and therefrom initiates simultaneously three equivalent propagating chains. Such a multi-site propagating mechanism seems to be consistent with the higher polymerization reaction rate observed in the Sc(OTf)3-catalysis. In comparison with other initiating systems, the rare-earth catalysis has the advantage of no requirement for microwave assistance, which would allow the whole polymerization process to be easily scaled up. We expect that the polyoxazolines bearing a chiral L-proline moiety in the side-chain synthesized herein will find potential applications in asymmetric catalysis, the related research is currently underway.Acknowledgements
The authors are indebted the financial support by the National Natural Science Foundation of China (Grant no. 21274122).Notes and references
- R. Hoogenboom, Macromol. Chem. Phys., 2007, 208, 18–25 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- H. Schlaad, C. Diehl, A. Gress, M. Meyer, A. L. Demirel, Y. Nur and A. Bertin, Macromol. Rapid Commun., 2010, 31, 511–525 CrossRef CAS PubMed.
- R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov and R. Jordan, Macromol. Rapid Commun., 2012, 33, 1613–1631 CrossRef CAS PubMed.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151–208 CrossRef CAS.
- C. Giardi, V. Lapinte, C. Charnay and J. J. Robin, React. Funct. Polym., 2009, 69, 643–649 CrossRef CAS PubMed.
- R. Luxenhofer, M. Bezen and R. Jordan, Macromol. Rapid Commun., 2008, 29, 1509–1513 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom, C. H. Abeln and U. S. Schubert, Macromol. Rapid Commun., 2004, 25, 1895–1899 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom, M. A. M. Leenen, M. A. R. Meier and U. S. Schubert, Macromolecules, 2005, 38, 5025–5034 CrossRef CAS.
- H. M. L. Lambermont-Thijs, H. P. C. V. Kuringen, J. P. W. V. D. Put, U. S. Schubert and R. Hoogenboom, Polymers, 2010, 2, 188–199 CrossRef CAS PubMed.
- K. Kempe, S. Jacobs, H. M. L. Lambermont-Thijs, M. M. W. M. Fijten, R. Hoogenboom and U. S. Schubert, Macromolecules, 2010, 43, 4098–4104 CrossRef CAS.
- M. M. Bloksma, U. S. Schubert and R. Hoogenboom, Polym. Chem., 2011, 2, 203–208 RSC.
- S. Cesana, J. Auernheimer, R. Jordan, H. Kessler and O. Nuyken, Macromol. Chem. Phys., 2006, 207, 183–192 CrossRef CAS.
- J.-S. Park, Y. Akiyama, F. M. Winnik and K. Kataoka, Macromolecules, 2004, 37, 6786–6792 CrossRef CAS.
- M. Bortenschlager, N. Schöllhorn, A. Wittmann, R. Weberskirch and R. Weberskirch, Chem.–Eur. J., 2007, 13, 520–528 CrossRef CAS PubMed.
- K. Kempe, S. L. Ng, K. F. Noi, M. Müllner, S. T. Gunawan and F. Caruso, ACS Macro Lett., 2013, 2, 1069–1072 CrossRef CAS.
- C. Legros, M.-C. De Pauw-Gillet, K. C. Tam, S. Lecommmandoux and D. Taton, Polym. Chem., 2013, 4, 4801–4808 RSC.
- Y. Isobe, D. Fujioka, S. Habaue and Y. Okamoto, J. Am. Chem. Soc., 2001, 123, 7180–7181 CrossRef CAS.
- Y. Isobe, Y. Suito, S. Habaue and Y. Okamoto, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1027–1033 CrossRef CAS.
- H. Baraki, S. Habaue and Y. Okamoto, Macromolecules, 2001, 34, 4724–4729 CrossRef CAS.
- K. Morioka, Y. Suito, Y. Isobe, S. Habaue and Y. Okamoto, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3354–3360 CrossRef CAS.
- W. Lu, L. P. Lou, F. Y. Hu, L. M. Jiang and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5411–5418 CrossRef CAS.
- G. X. Liu, W. Lu, L. M. Jiang, W. L. Sun and Z. Q. Shen, Acta Polym. Sin., 2009, 775–780 CrossRef.
- J. Ling, Z. Q. Shen and Q. H. Huang, Macromolecules, 2001, 34, 7613–7616 CrossRef CAS.
- J. Ling, Y. F. Zhang and Z. Q. Shen, Chin. Chem. Lett., 2001, 12, 41–42 CAS.
- L. X. You, T. E. Hogen-Esch, Y. H. Zhu, J. Ling and Z. Q. Shen, Polymer, 2012, 53, 4112–4118 CrossRef CAS PubMed.
- H. Peng, J. Ling and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1076–1085 CrossRef CAS.
- T. Kagiya, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 441–445 CrossRef CAS.
- S. Kobayashi and I. Hachiya, J. Org. Chem., 1994, 59, 3590–3596 CrossRef CAS.
- J. Lustoň, J. Kronek and F. Böhme, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 343–355 CrossRef.
- K. Hioki, Y. Takechi, N. Kimura, H. Tanaka and M. Kunishima, Chem. Pharm. Bull., 2008, 56, 1735–1737 CrossRef CAS.
- M. T. Leffler and R. Adams, J. Am. Chem. Soc., 1937, 59, 2252–2258 CrossRef CAS.
- M. M. Bloksma, S. Rogers, U. S. Schubert and R. Hoogenboom, Soft Matter, 2010, 6, 994–1003 RSC.
- R. Hoogenboom, M. A. M. Leenen, F. Wiesbrock and U. S. Schubert, Macromol. Rapid Commun., 2005, 26, 1773–1778 CrossRef CAS.
- A. Baumgaertel, C. Weber, K. Knop, A. Crecelius and U. S. Schubert, Rapid Commun. Mass Spectrom., 2009, 23, 756–762 CrossRef CAS PubMed.
- Q. Liu, M. Konas and J. S. Riffle, Macromolecules, 1993, 26, 5572–5576 CrossRef CAS.
- L. X. You, Z. Q. Shen, J. Kong and J. Ling, Polymer, 2014, 55, 2404–2410 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1026078. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra11404c |
| This journal is © The Royal Society of Chemistry 2014 |