
Poly(4-methyl-1-pentene)/alkylated graphene oxide nanocomposites: the emergence of a new crystal structure
Abstract
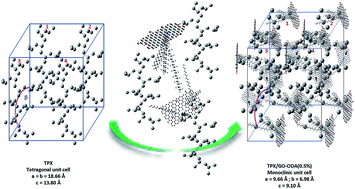
Modified graphene oxide (GO) not only offers a wrinkled structure but may also transform the crystal structure, giving it a potential application in high performance polymer/filler composites. In this work, alkylated graphene oxide (GO–ODA) is obtained via mutual electrostatic interaction between the epoxide group of GO and the amine group of octadecylamine (ODA). Then nanocomposites are obtained via introducing different contents of GO–ODA to poly(4-methy-1-pentene) (PMP or TPX) using a solution approach. It is found that the obtained GO–ODA has no effect on the crystallinity of TPX. It is worth noting that GO–ODA changes the crystal structure. GO, as a kind of nanofiller, is used to change the crystal structure of polymer/filler composites for the first time, which is an important finding and obviously provides a good example to widen the application of GO.
 Please wait while we load your content...
Please wait while we load your content...

