
A joint experimental and theoretical study on the effect of manganese doping on the structural, electrochemical and physical properties of lithium iron phosphate
Abstract
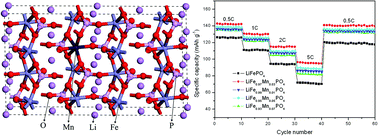
Mn doped LiFe1−xMnxPO4 (x = 0, 0.01, 0.03, 0.05, 0.07) cathode materials are synthesized by a carbothermal reduction method. The structure, morphology and electrochemical performance of the samples are characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), Raman, and charging–discharging tests. It is found that an appropriate amount of Mn doping did not affect the olivine structure and morphology of LiFePO4, but greatly improves its electrochemical performance. Especially, the LiFe0.97Mn0.03PO4 sample exhibits the best electrochemical performance, which shows the maximum discharge capacity of 148.2 mA h g−1 at 0.1 C and a capacity retention ratio of 98% after 60 cycles at various rates. Based on first-principles density functional theory (DFT), the lithium ion migration channels, energy band structures, densities of states, and charge density diffusion of LiFePO4 and LiFe15/16Mn1/16PO4 are calculated to further investigate the influence of Mn doping on the LiFePO4 lattice. The results prove that the higher lithium ion conduction and electronic conductivity of LiFe15/16Mn1/16PO4 are attributed to Mn doping.
 Please wait while we load your content...
Please wait while we load your content...

