DOI:
10.1039/C4RA11331D
(Paper)
RSC Adv., 2014,
4, 63442-63446
The crystal structure of IrB2: a first-principle calculation
Received
29th October 2014
, Accepted 10th November 2014
First published on 10th November 2014
Abstract
First-principle calculations were performed to investigate the structural, elastic, and electronic properties of iridium diboride (IrB2). It was demonstrated that the new phase of IrB2 belongs to the monoclinic C2/m space group, and we have named it m-IrB2. Its structure is energetically much superior to the recently proposed Pmmn-type IrB2. Further calculations of phonon and elastic constants confirm that m-IrB2 is dynamically and mechanically stable. The calculated high shear modulus reveals that it is a potentially a material of low compressibility. An analysis of the density of its states and chemical bonding show that the strongly directional covalent B–B and B–Ir bonds in m-IrB2 make a considerable contribution to its stability.
1. Introduction
Any material with Vickers hardness HV above 40 GPa may be defined as a superhard material.1 Superhard materials are of considerable interest, not only for their hardness, but also because they are ultra-incompressible, and they have high melting temperature, chemical inertness, and high thermal conductivity, and thus, they are important for industrial applications.2 The most widely used superhard materials are diamond3 and cubic boron nitride (c-BN),4 although the application of these is limited by their natural defects. Finding novel, improved superhard materials is therefore of high priority.
The main principle in the design of hard materials is to combine small strongly covalent bonding light elements, such as boron, carbon, or nitrogen, with large electron-rich transition metals. These compounds usually exhibit high valence electron density and highly directional covalent bonding, making them a rich source of multifunctional superhard materials.5–9 It has been reported that ReB2,10,13 OsB2 (ref. 11 and 13) and RuB2 (ref. 12 and 13) have been identified as superhard materials, and theoretical studies support these results. ReB2 is particularly interesting. OsB2 and ReB2 form different lattice structures: OsB2 has an orthorhombic RuB2-type structure (space group Pmmn), and ReB2 crystallizes in a hexagonal ReB2-type structure (space group P63/mmc).15–17 The hardness of ReB2 is 48 GPa at a load of 0.49 N.14 Could the other 5d transition metal borides also present such an interesting nature? Ir and Re are neighboring 5d transition metals, and a pure-phase IrB1.35 has been produced by X-ray diffraction. The Vickers hardness of IrB1.35 is 18.2–49.8 GPa, depending on the load, ranging from 0.49 to 9.81 N.18 In addition, a superhard IrB1.1 film, with a high Vickers hardness of 43 GPa has also been found.19,20 Recently, Wang et al. have reported that IrB2 in the orthorhombic phase, with a Pmmn space group, was most stable at ambient pressures. According to this study, the bulk modulus and shear modulus of IrB2 were 277 GPa and 108 GPa, respectively.21 IrB2 is thus a good candidate as a superhard material, and it was hoped that a study of its high-pressure structures and phase transitions might amplify the understanding of transition metal compounds and their behavior.
In the present study the structural stability, elastic properties, and electronic structure of iridium diboride (IrB2) using first-principle calculations based on density functional theory have been explored. A monoclinic C2/m structure IrB2 has been revealed which is much more energetically preferable to the Pmmn-type structures recently proposed, and it has been named it m-IrB2. The new phase is dynamically and mechanically stable. The phase stability, electronic structure, and elastic properties of different structures have been compared and analyzed in detail. The quadrilateral networks of the boron layers provide strong covalent bonding, a prerequisite for a hard material of low compressibility.
2. Computational details
In the present study evolutionary variable-cell simulations for IrB2 were performed at ambient pressure, involving systems containing one to four formula units (FU), as implemented in the USPEX code.22–24 Most calculations were performed using first-principle plane-wave pseudo-potential density functional theory according to the Vienna ab initio simulation package (VASP).25–27 The exchange and correlation effects are described by the local-density approximation.28 The k-point sampling in the Brillouin zone (BZ) was performed using the Monkhorst–Pack scheme. For hexagonal structures Γ-centered grids were used. An energy cut-off at 500 eV with 4 × 9 × 5, 6 × 8 × 7, 5 × 8 × 3, 4 × 4 × 9 and 8 × 5 × 6 Monkhorst–Pack grids for the electronic BZ integrations was conducted for the m-IrB2, C2/m-IrB2, Cm-IrB2, Pnnm-IrB2, and Pmmn-IrB2 phases, respectively. Formation enthalpy, ΔH, was calculated using eqn (1):| | |
ΔH = E(IrB2) − E(solid Ir) − 2E(solid B),
| (1) |
where the symbol E represents one FU of total energy for each solid phase, the solid phase of boron being the α-phase. All these were chosen to ensure that enthalpy calculations were fully convergent, with energy differences below 1 meV per atom. For each selected structure the bond lengths, cell parameters, and atomic positions, were fully optimized at different pressures. Elastic constants were obtained by evaluation of the stress tensor generated by small strains, using the density-functional plane wave technique used in the CASTEP code.29 The bulk modulus, shear modulus, Young's modulus, and Poisson's ratio were estimated using the Voigt–Reuss–Hill approximation.30 The phonon calculations were carried out using a super-cell approach, as implemented in the Phonon code.31 The theoretical Vickers hardness was estimated using the Chen,32 Gao,33 and Šimůnek models.34 Details of the convergence tests are described elsewhere.35–38
3. Results and discussion
3.1 Structure and features
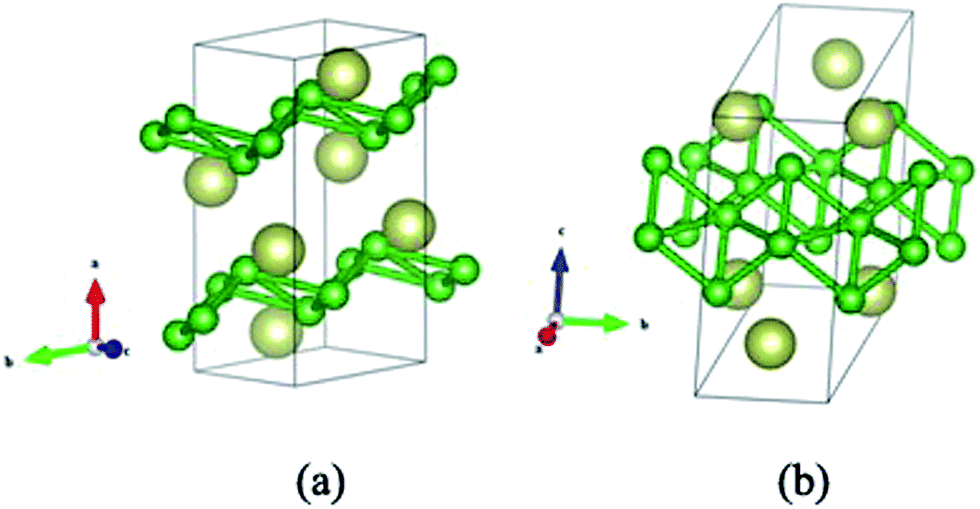
At ambient pressure variable cell structure prediction simulations were performed for IrB2 with a system containing one to four FUs in the simulation cell. The variable cell simulation showed the monoclinic C2/m structure to be the most stable phase, as illustrated in Fig. 1. There are 12 atoms in the unit cell. The m-IrB2 unit cell is a monoclinic structure (space group, C2/m) with a = 7.346 Å, b = 2.810 Å and c = 5.837 Å, α = γ = 90°, β = 67.22° and one non-equivalent Ir atom occupies the crystallographic 4i (0.409, 0.000, 0.836) sites and two non-equivalent B atoms occupy the crystallographic 4i (0.669, 0.000, 0.488), and (0.768, 0.5, 0.264) sites at zero pressure. In m-IrB2, the boron layers form a type of quadrilateral conjugated network, and the Ir atoms are arranged in corrugated hexagonal sheets perpendicular to the c axis. The shortest inter-atomic distance of B–Ir is 2.18 Å, which is less than the sum (2.25 Å) of the covalent radii of the B atom (r = 0.84 Å) and the Ir atom (r = 1.41 Å), suggesting strong covalent bonding.
 |
| | Fig. 1 Crystal structure of m-IrB2 in different directions. | |
3.2 Thermodynamic and dynamic stability
The application of a compound requires an accurate knowledge of its thermodynamic stability, and in particular its phase stability. For further experimental synthesis it was therefore necessary to investigate the relative stability of IrB2. The formation enthalpies were evaluated using eqn (2):| | |
H = E(IrB2) − E(Ir) − 2E(B),
| (2) |
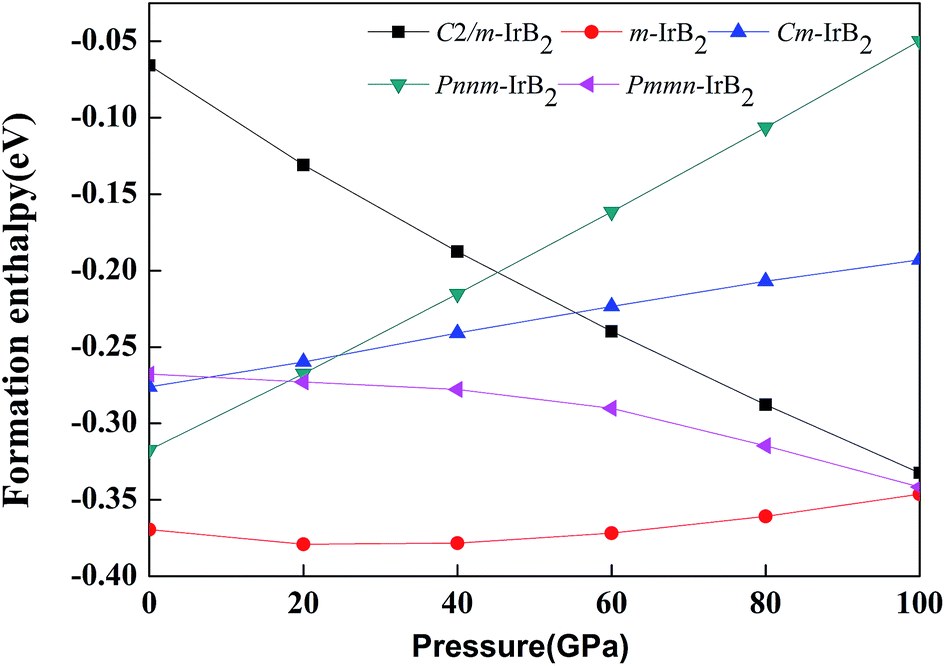
where E(B) was calculated from the structure of α-rhombohedral boron.39 Fig. 2 illustrates the calculated formation enthalpy of IrB2 with different structures under pressures up to 100 GPa. According to these enthalpy calculations, only C2/m-IrB2, Cm-IrB2, Pnnm-IrB2, and m-IrB2 were competitive structures, and Wang's Pmmn-IrB2 structure is also included in Fig. 2.
 |
| | Fig. 2 Formation enthalpy versus pressure for different IrB2 structures. | |
As can be seen in Fig. 2, all the structures had a negative formation enthalpy with thermodynamic stability below 100 GPa. With increase in pressure the stability of Pnnm-IrB2, Cm-IrB2, and m-IrB2 gradually decreased, whereas the stability of the other two phases increased. At ambient pressure, the formation enthalpy predicted for m-IrB2 was much lower than that of the reactants, Ir + 2B, by a factor of 0.37 eV, indicating that it should be directly synthesizable from elemental Ir and B. In Wang's paper, Pmmn-IrB2 is stated to be the most stable structure, but the monoclinic m-IrB2 structure is in fact more stable than this. With increasing pressure the enthalpy of m-IrB2 becomes close to that of Pmmn-IrB2. At least up to 100 GPa, the enthalpy of m-IrB2 is in all cases the lowest of the candidate structures.
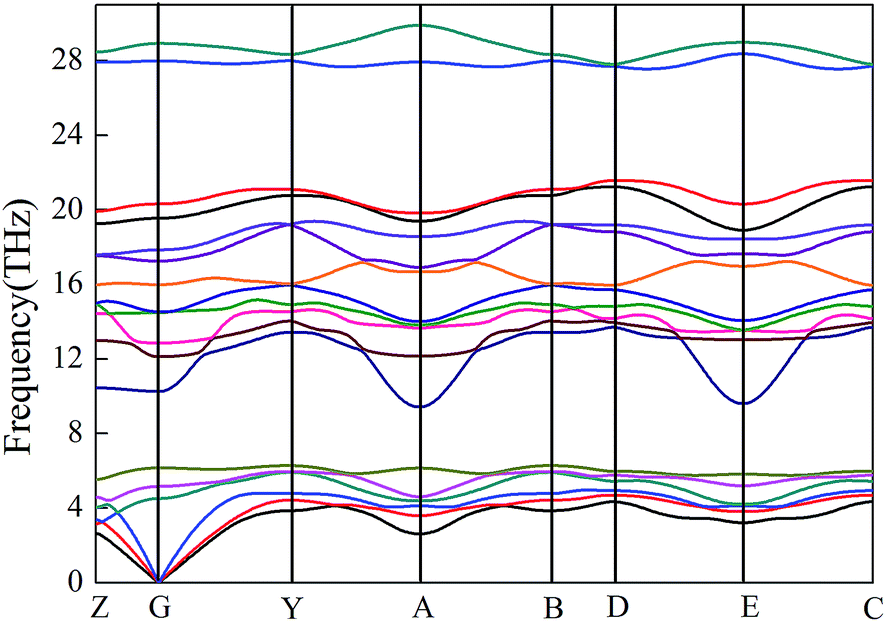
Dynamic stability is important for structural stability. Phonon structures provide a criterion for judging crystal stability. The phonon–dispersion curves in the whole BZ of m-IrB2 at ambient conditions are shown in Fig. 3, which shows that there were no imaginary phonon frequencies in the whole BZ, confirming the dynamic stability of the newly proposed m-IrB2. It is well known that shorter bond lengths contribute to higher phonon frequencies. The phonon frequency of m-IrB2 (∼30 THz) shown in Fig. 3 was larger than that of Wang's Pmmn-IrB2 (∼24 THz), and this high phonon frequency indicates that there are shorter bond lengths in m-IrB2.
 |
| | Fig. 3 Phonon–dispersion curves of m-IrB2 at 0 GPa. | |
3.3 Mechanical stability
The elastic constants were calculated in order to evaluate the mechanical properties and stability of m-IrB2. The mechanical stability of a crystal requires the strain energy to be positive, which implies that the whole set of elastic constant, Cij, values satisfies the Born–Huang criterion.40 For a monoclinic crystal, the independent elastic stiffness tensor comprises thirteen components: C11, C22, C33, C44, C55, C66, C12, C13, C23, C15, C25, C35, and C46. The criteria for mechanical stability are given by eqn (3) and (4):| | |
C11 > 0, C22 > 0, C33 > 0, C44 > 0, C55 > 0, C66 > 0, (C33C55 − C352) > 0, (C44C66 − C462) > 0, (C22 + C33 − 2C23) > 0, [C22(C33C55 − C352) + 2C23C25C35 − C232C55 − C252 − C33] > 0,
| (3) |
| | |
g = C11C22C33 − C11C232 − C22C132 − C33C122 + 2C12C13C23, {2[C15C25(C33C12 − C13C23) + C15C35(C22C13 − C12C33) + C25C35(C11C23 − C12C13)] − [C152(C22C33 − C232) + C252(C11C33 − C132) + C352(C11C22 − C122)] + C55g} > 0
| (4) |
From Table 1 we can confirm that the Cij of m-IrB2 fulfills the stability criteria listed previously, suggesting that m-IrB2 is mechanically stable at ambient pressure. It is therefore apparent that m-IrB2 is both mechanically and dynamically stable at ambient pressure. Accurate elastic constants are helpful in understanding mechanical properties, and they also provide vital information in predicting the properties of a material. To explore the mechanical properties of m-IrB2 the elastic constants of m-IrB2, Pmmn-IrB2, C2/m-IrB2, Cm-IrB2, and Pnnm-IrB2 were calculated and these are summarized in Table 1. In addition, the Young's modulus, Y, and Poisson's ratio, υ, were obtained from eqn (5) and (6):
| | |
υ = (3B − 2G)/(6B + 2G)
| (6) |
Table 1 Calculated elastic constants, Cij (GPa), bulk modulus, B (GPa), shear modulus, G (GPa), Young's modulus, Y (GPa), Poisson's ratio, υ, Vicker's hardness, Hv and the B/G ratio, for m-IrB2, Pmmn-IrB2, C2/m-IrB2, Cm-IrB2, and Pnnm-IrB2
| |
C11 |
C22 |
C33 |
C44 |
C55 |
C66 |
C12 |
C13 |
C23 |
B |
G |
Y |
υ |
B/G |
Hv |
| m-IrB2 |
586 |
556 |
816 |
136 |
201 |
78 |
178 |
311 |
129 |
355 |
172 |
444 |
0.29 |
2.06 |
13.82 |
| Pmmn-IrB2 |
369 |
429 |
670 |
45 |
70 |
155 |
246 |
151 |
174 |
290 |
113 |
300 |
0.32 |
2.56 |
7.20 |
| C2/m-IrB2 |
650 |
480 |
674 |
179 |
137 |
91 |
226 |
285 |
252 |
370 |
151 |
398 |
0.32 |
2.45 |
9.70 |
| Cm-IrB2 |
549 |
453 |
864 |
119 |
179 |
190 |
255 |
222 |
189 |
355 |
177 |
455 |
0.28 |
2.0 |
14.68 |
| Pnnm-IrB2 |
553 |
761 |
507 |
127 |
107 |
224 |
346 |
142 |
150 |
344 |
170 |
437 |
0.28 |
2.02 |
14.02 |
The calculated bulk modulus, shear modulus, Young's modulus, and Poisson's ratio of m-IrB2 and the reference materials mentioned previously are summarized in Table 1.
As shown in Table 1, the calculated C33 for m-IrB2 was larger than that for C11 or C22, implying that the resistance to deformation in the c-direction is stronger than in the a- or the b-direction. The calculated bulk modulus of the m-IrB2 phase was 355 GPa, which is close to the experimental data for OsB2 (348 GPa),11 indicating its ultra-incompressible structure. It is well known that shear modulus provides a better indicator of potential hardness than bulk modulus. The calculated shear moduli of m-IrB2, Pmmn-IrB2, C2/m-IrB2, Cm-IrB2, and Pnnm-IrB2 are 172, 113, 151, 177 and 170 GPa, respectively. It is expected that they will resist shear strain to a large extent. The high Young's moduli of these compounds indicate their ability to resist tension and pressure in the range of elastic deformation.41
The B/G ratio describes the ductility or brittleness of a material, with 1.75 as the critical value. If the value of B/G lies below 1.75 the material is brittle, otherwise it is ductile.42 The calculated B/G ratio of m-IrB2 was 2.06, implying a ductile nature. The value of Poisson's ratio (υ) indicates the directionality of the covalent bonding. A typical υ value for covalent materials is 0.1 and for metallic materials it is 0.33.43 A small Poisson's ratio contributes to the hardness of a material and indicates a high degree of covalent bonding. Chen's model was used for calculating the hardness:
| | |
Hv = 2(K2G)0.585 − 3,
| (7) |
where
K =
G/
B is Pugh's modulus ratio. Using this model the hardness of m-IrB
2 was calculated to be 14.4 GPa. The calculated hardness was 12.05 GPa using Gao's model and 15.03 GPa using the Šimůnek model. These results, show the value of Vickers hardness by different empirical hardness models lies in the range 12.05–15.03 GPa, thus confirming that m-IrB2 is indeed a hard material, with a mean hardness value of ∼13.82 GPa.
3.4 Electronic structure analysis
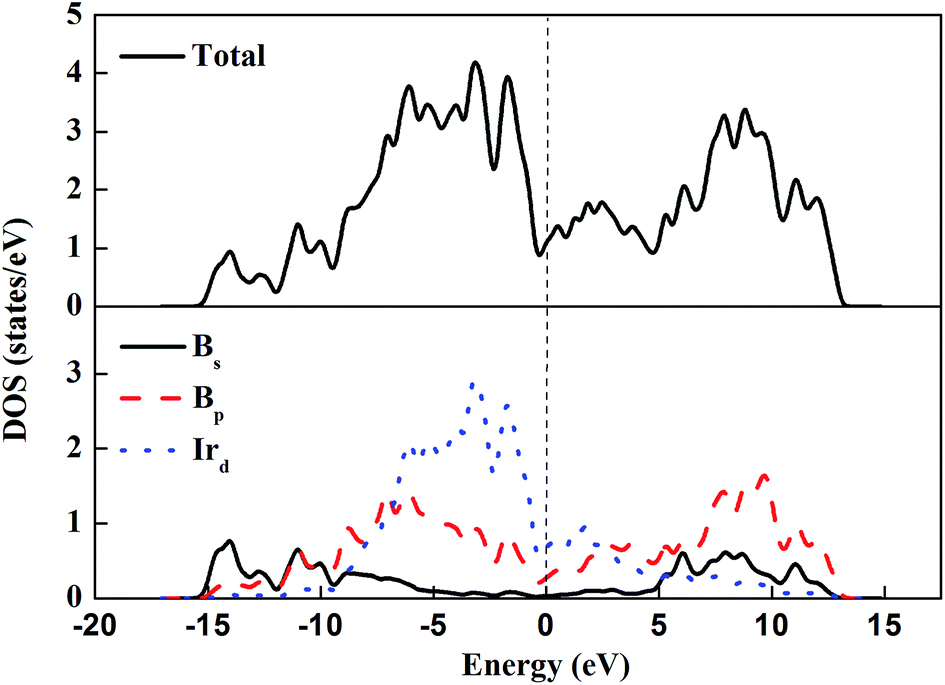
The electronic density of states (DOS) and the atom-resolved partial density of states (PDOS) of m-IrB2 are shown in Fig. 4. An intriguing type of bonding is seen in the PDOS of m-IrB2, in which the iridium and boron atoms form strong covalent bonds, which is confirmed by the appreciable overlap of the iridium d-electron and the boron p-electron curves. From the PDOS of m-IrB2 it is seen that the iridium d-electron states contribute most to the DOS, and the boron s-electron states are mainly located at the bottom of the valence bands. The finite electronic DOS at the Fermi level indicates the metallic nature of IrB2 under ambient pressure.
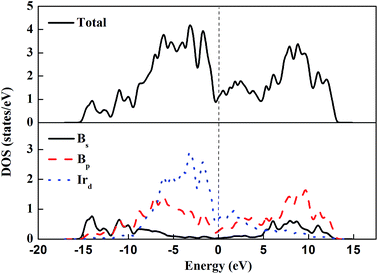 |
| | Fig. 4 Total and partial DOS of m-IrB2. The Fermi level is indicated by a dashed line. | |
To gain a more detailed insight into the bonding character of m-IrB2 the electronic localization function (ELF) was calculated.44 The ELF provides a reliable measure of electron pairing and localization. According to its original definition, ELF values lie between 0 and 1, where ELF = 1 corresponds to the perfect localization characteristic of covalent bonds. It should be noted that ELF is not a measure of electron density but is a function of the Pauli principle, and is valuable for distinguishing between metallic, covalent, and ionic bonding. To determine the chemical bonding of m-IrB2 we plotted its ELF in Fig. 5, with the iso-surface at ELF = 0.74. In addition, strong covalent bonding interaction between the B atoms is seen in m-IrB2. This is consistent with analysis of the DOS, and as a result this strong covalent bonding will naturally increase the structural stability and high bulk modulus of m-IrB2.
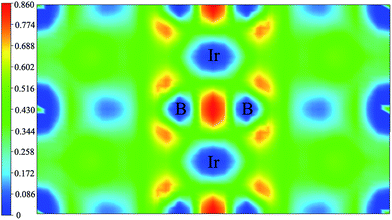 |
| | Fig. 5 Contours of the electronic localization function (ELF) of m-IrB2. | |
4. Conclusions
A monoclinic C2/m structure has been shown to be the ground-state structure of IrB2, using first-principle calculations based on density functional theory. It was proposed that it be named m-IrB2 and this structure is energetically preferable to the proposed Pmmn structure suggested earlier. The structural stability of IrB2 was reviewed, and up to pressures of at least 100 GPa, the enthalpy of m-IrB2 is always the lowest of the candidate structures. According to the calculated phonon dispersions and elastic constants of m-IrB2, it is concluded that this phase is both dynamically and elastically stable under ambient conditions. Its high bulk and shear moduli imply that m-IrB2 is potentially a non-compressible and superhard material. In addition, the calculated PDOS results demonstrate that m-IrB2 is metallic in character. The calculated electronic localization function demonstrates that strongly covalent B–B and Ir–B bonding plays a major role in establishing the incompressibility and hardness of m-IrB2.
Acknowledgements
We are grateful for financial support from the National Basic Research Program of China (no. 2011CB808200), the Program for Changjiang Scholars and Innovative Research Team in University (no. IRT1132), the National Natural Science Foundation of China (no. 51032001, 11074090, 11404134, 10979001, 51025206, 11104102, 11204100), the National Fund for Fostering Talent in Basic Science (no. J1103202), and the China Postdoctoral Science Foundation (2014M561279, 2013T60314, 2012M511326). Specialized Research Fund for the Doctoral Program of Higher Education (20110061120007, 20120061120008). Some of the calculations were carried out at the High Performance Computing Center (HPCC) of Jilin University.
References
- J. Haines, J. M. Léger and G. Bocquillon, Annu. Rev. Mater. Res., 2001, 31, 1–23 CrossRef CAS.
- B. L. Jonathan, H. T. Sarah and B. K. Richard, Adv. Funct. Mater., 2009, 19, 3519–3533 CrossRef.
- F. Occelli, D. L. Farber and R. L. Toullec, Nat. Mater., 2003, 2, 151–154 CrossRef CAS PubMed.
- J. C. Zheng, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 052105 CrossRef.
- A. L. Ivanovskii, Prog. Mater. Sci., 2012, 57, 184–228 CrossRef CAS PubMed.
- R. W. Cumberland, M. B. Weinberger, J. J. Gilman, S. M. Clark, S. H. Tolbert and R. B. Kaner, J. Am. Chem. Soc., 2005, 127, 7264–7265 CrossRef CAS PubMed.
- X. F. Hao, Y. H. Xu, Zh. J. Xu, D. F. Zhou, X. J. Liu, X. Q. Cao and J. Meng, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 224112 CrossRef.
- H. Y. Chung, M. B. Weinberger, J. M. Yang, S. H. Tolbert and R. B. Kaner1, Appl. Phys. Lett., 2008, 92, 261904 CrossRef PubMed.
- Z. S. Zhao, M. Wang, L. Cui, J. L. He, D. L. Yu and Y. J. Tian, J. Phys. Chem. C, 2010, 114, 9961–9964 CAS.
- H. Y. Chung, M. B. Weinberger, J. B. Levine, A. Kavner, J. M. Yang, S. H. Tolbert and R. B. Kaner, Science, 2007, 316, 436–439 CrossRef CAS PubMed.
- Z. Y. Chen, H. J. Xiang, J. L. Yang, J. G. Hou and Q. Sh. Zhu, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 012102 CrossRef.
- Y. Ch. Wang, T. K. Yao, L. M. Wang, J. L. Yao, H. Li, J. W. Zhang and H. Y. Gou, Dalton Trans., 2013, 42, 7041–7050 RSC.
- Q. F. Gu, G. Krauss and W. Steurer, Adv. Mater., 2008, 20, 3620 CrossRef CAS.
- J. Q. Qin, D. W. He, J. H. Wang, L. M. Fang, L. Lei, Y. J. Li, J. Hu, Z. L. Kou and Y. Bi, Adv. Mater., 2008, 20, 4780–4783 CrossRef CAS.
- Y. X. Wang, Appl. Phys. Lett., 2007, 91, 101904 CrossRef PubMed.
- X. Q. Chen, C. L. Fu, M. Krcmar and G. S. Painter, Phys. Rev. Lett., 2008, 100, 196403 CrossRef.
- J. Wang and Y. J. Wang, J. Appl. Phys., 2009, 105, 083539 CrossRef PubMed.
- J. V. Rau and A. Latini, Chem. Mater., 2009, 21, 1407–1409 CrossRef CAS.
- A. Latini, J. V. Rau, R. Teghil, A. Generosi and V. R. Albertini, ACS Appl. Mater. Interfaces, 2010, 2, 581–587 CAS.
- Y. Pan, W. T. Zheng, X. Y. Hu, L. Qiao and S. Chen, J. Alloys Compd., 2014, 587, 468–473 CrossRef CAS PubMed.
- D. Y. Wang, B. Wang and Y. X. Wang, J. Phys. Chem. C, 2012, 116, 21961–21966 CAS.
- A. R. Oganov and C. W. Glass, J. Chem. Phys., 2006, 124, 244704 CrossRef PubMed.
- A. R. Oganov, A. O. Lyakhov and M. Valle, Acc. Chem. Res., 2011, 44, 227–237 CrossRef CAS PubMed.
- A. O. Lyakhov, A. R. Oganov, H. T. Stokes and Q. Zhu, Comput. Phys. Commun., 2013, 184, 1172–1182 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. J. Hafner, Phys. Condens. Matter, 1994, 6, 8245 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- D. M. Ceperley and B. J. Alder, Phys. Rev. Lett., 1980, 45, 566 CrossRef CAS.
- M. D. Segall, P. L. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- R. Hill, Proc. Phys. Soc., London, Sect. A, 1952, 65, 349–354 CrossRef.
- K. Parlinski, Computer code PHONON, http://wolf.ifj.edu.pl/phonon/.
- X. Q. Chen, H. Y. Niu, D. Z. Li and Y. Y. Li, Intermetallics, 2011, 19, 1275–1281 CrossRef CAS PubMed.
- F. M. Gao, J. L. He, E. D. Wu, S. M. Liu, D. L. Yu, D. C. Li, S. Y. Zhang and Y. J. Tian, Phys. Rev. Lett., 2003, 91, 015502 CrossRef.
- A. Šimůnek, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 172108 CrossRef.
- D. Li, K. Bao, F. B. Tian, Z. W. Zeng, Z. He, B. B. Liu and T. Cui, Phys. Chem. Chem. Phys., 2012, 14, 4347–4350 RSC.
- D. Li, F. B. Tian, D. F. Duan, K. Bao, B. H. Chu, X. J. Sha, B. B. Liu and T. Cui, RSC Adv., 2014, 4, 10133–10139 RSC.
- D. Li, F. B. Tian, D. F. Duan, Z. L. Zhao, Y. X. Liu, B. H. Chu, X. J. Sha, L. Wang, B. B. Liu and T. Cui, RSC Adv., 2014, 4, 17364–17369 RSC.
- B. H. Chu, D. Li, K. Bao, F. B. Tian, D. F. Duan, X. J. Sha, P. G. Hou, Y. X. Liu, H. D. Zhang, B. B. Liu and T. Cui, J. Alloys Compd., 2014, 617, 660–664 CrossRef CAS PubMed.
- A. R. Oganov, J. H. Chen, C. L. Gatti, Y. M. Ma, C. W. Glass, Z. X. Liu, T. Yu, O. O. Kurakevych and V. L. Solozhenko, Nature, 2009, 457, 863 CrossRef CAS PubMed.
- Z. J. Wu, E. J. Zhao, H. P. Xiang, X. F. Hao, X. J. Liu and J. Meng, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 054115 CrossRef.
- M. Born, Proc. Cambridge Philos. Soc., 1940, 36, 160 CrossRef CAS.
- S. F. Pugh, Philos. Mag., 1954, 45, 823–843 CAS.
- J. Haines, J. M. Leger and G. Bocquillon, Annu. Rev. Mater. Res., 2001, 31, 1–23 CrossRef CAS.
- A. Savin, H. J. Flad, J. Flad, H. Preuss and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1992, 31, 185–187 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.