
Magnetic, X-ray and Mössbauer studies on magnetite/maghemite core–shell nanostructures fabricated through an aqueous route†
Srividhya J. Iyengara,
Mathew Joya,
Chandan Kumar Ghoshb,
Subhrajyoti Deyc,
Ravinder K. Kotnalad and
Swapankumar Ghosh*a
aAdvanced Clay & Traditional Ceramics Division, CSIR-Central Glass & Ceramics Research Institute, Kolkata-700032, India. E-mail: swapankumar.ghosh2@mail.dcu.ie; srividhyaji@gmail.com; Fax: +91 33 24730957; Tel: +91 33 23223546
bSchool of Material Science & Nanotechnology, Jadavpur University, Kolkata-700032, India
cDepartment of Physics, Jadavpur University, Kolkata-700032, India
dMaterials Physics and Engineering, CSIR-National Physical Laboratory, New Delhi-110 012, India
First published on 18th November 2014
Abstract
Uniform 6–13 nm sized 0D superparamagnetic Fe3O4 nanocrystals were synthesized by an aqueous ‘co-precipitation method’ under a N2 atmosphere as a function of temperature to understand the growth kinetics. The crystal phases, surface charge, size, morphology and magnetic characteristics of as-synthesized nanocrystals were characterized by XRD, Raman spectroscopy, FTIR, TG-DTA, BET surface area, dynamic light scattering along with zeta potential, HR-TEM, EDAX, vibrating sample magnetometry and Mössbauer spectroscopy. TEM investigation revealed highly crystalline spherical magnetite particles in the 8.2–12.5 nm size range. The kinetically controlled as-grown nanoparticles were found to possess a preferential (311) orientation of the cubic phase, with a highest magnetic susceptibility of ∼57 emu g−1. The Williamson–Hall technique was employed to evaluate the mean crystallite size and microstrain involved in the as-synthesized nanocrystals from the X-ray peak broadening. In addition to FTIR and Raman spectra, Rietveld structural refinement of XRD confirms the magnetite phase with 5–20% maghemite in the sample. VSM and Mössbauer spectral data allowed us to fit the magnetite/maghemite content to a core–shell model where the shell is 0.2–0.3 nm thick maghemite over a magnetite core. The activation energy of <10 kJ mol−1 calculated from an Arrhenius plot for the complex process of nucleation and growth by diffusion during synthesis shows the significance of the precipitation temperature in the size controlled fabrication processes of nanocrystals. Brunauer–Emmett–Teller (BET) results reveal a mesoporous structure and a large surface area of 124 m2 g−1. Magnetic measurement shows that the particles are ferromagnetic at room temperature with zero remanence and zero coercivity. This method produced highly crystalline and dispersed 0D magnetite nanocrystals suitable for biological applications in imaging and drug delivery.
Introduction
Magnetite (Fe3O4, containing Fe2+ and Fe3+ in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio), crystallizes in the inverse cubic spinel structure Fd
:2 ratio), crystallizes in the inverse cubic spinel structure Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m above the so-called Verwey transition temperature ∼120 K.1 The oxygen atoms form the close-packed face-centered-cubic (fcc) lattices with the iron atoms occupying the interstitial positions.2 Fe3O4 nanoparticles exhibit unique and tunable fundamental size- and shape-dependent novel magnetic, optical, and other unique properties due to quantum confinement effects i.e., the nanometer size effect and have attracted great attention in recent years. The crystal chemistry of Fe3O4 is of considerable interest to mineralogists and materials scientists because of its extensive applications in magnetic resonance imaging (MRI) for imaging the brain and the central nervous system,3–5 for assessing cardiac function,6 as a drug delivery/gene-delivery platform,7 ferro-fluids, MICR ink,8 cell targeting,9 magnetic force based tissue engineering,10,11 magnetically controllable catheters, glucose sensing,12 sensing tumor by magneto-impedance,13 and magnetic separation of biological materials. Outstanding magneto-electrical properties have also been reported in strain induced magnetite.14,15 Recent technological advances in smart multifunctional nanobiomagnetic platforms offer exciting opportunities in personalized medicine for more accurate early prognosis, monitoring and treatment of various diseases without jeopardizing healthy tissues by identifying unique biochemical markers of disease before the appearance of symptoms obviates the need.16 Nanocrystalline Fe3O4 often exhibits superparamagnetic behavior. Néel relaxation of superparamagnetic Fe3O4 is an effective way to heat up the nanocrystals and the surrounding tissue by transferring energy from the external magnetic field in therapeutic hyperthermia and, more recently, in the development of theranostic technologies.1,17
m above the so-called Verwey transition temperature ∼120 K.1 The oxygen atoms form the close-packed face-centered-cubic (fcc) lattices with the iron atoms occupying the interstitial positions.2 Fe3O4 nanoparticles exhibit unique and tunable fundamental size- and shape-dependent novel magnetic, optical, and other unique properties due to quantum confinement effects i.e., the nanometer size effect and have attracted great attention in recent years. The crystal chemistry of Fe3O4 is of considerable interest to mineralogists and materials scientists because of its extensive applications in magnetic resonance imaging (MRI) for imaging the brain and the central nervous system,3–5 for assessing cardiac function,6 as a drug delivery/gene-delivery platform,7 ferro-fluids, MICR ink,8 cell targeting,9 magnetic force based tissue engineering,10,11 magnetically controllable catheters, glucose sensing,12 sensing tumor by magneto-impedance,13 and magnetic separation of biological materials. Outstanding magneto-electrical properties have also been reported in strain induced magnetite.14,15 Recent technological advances in smart multifunctional nanobiomagnetic platforms offer exciting opportunities in personalized medicine for more accurate early prognosis, monitoring and treatment of various diseases without jeopardizing healthy tissues by identifying unique biochemical markers of disease before the appearance of symptoms obviates the need.16 Nanocrystalline Fe3O4 often exhibits superparamagnetic behavior. Néel relaxation of superparamagnetic Fe3O4 is an effective way to heat up the nanocrystals and the surrounding tissue by transferring energy from the external magnetic field in therapeutic hyperthermia and, more recently, in the development of theranostic technologies.1,17
Large surface area to volume ratio for nanoparticles provides enormous driving force for diffusion, especially at elevated temperatures.18 Colombo et al. calculated the activation energy for the reduction of iron oxides (hematite, maghemite and magnetite) at 150° to 400 °C under reductive atmosphere.19 Brus et al. reported the diffusion controlled aqueous oxidation kinetics of magnetite nanoparticles.20 Although magnetite nanocrystals prepared by the precipitation technique have been extensively studied, to the best of our knowledge, seldom efforts have been devoted on its growth kinetics during crystallisation. Magnetite NPs are susceptible to undergo surface oxidation in air to maghemite (γ-Fe2O3), and hence the control of magnetic properties at these very small sizes still remains a challenge. Average size estimation by transmission electron microscopy (TEM) is by analyzing rather a limited number of nanoparticles (typically 100–300) compared to >1012 NPs investigated by X-ray diffraction (XRD) and vibrating sample magnetometry (VSM).21 Debye–Scherrer formula, though widely been used to estimate crystal sizes, underestimates the grain size as it ignores the line broadening due to microstrain in the lattice as a result packing defects/dislocation structures etc.22 Application of Williamson–Hall (W–H) method on X-ray profiles not only estimate the crystallite sizes but also isolates the peak broadening due to internal lattice strain.23
Over the past couple of decades numerous techniques have been proposed to synthesize nano-sized Fe3O4 particles, such as sol–gel,24 hydrothermal/solvothermal1,25 thermal decomposition,26 reverse micelles,27 polyol,28 sonolysis,29 gamma ray irradiation,30 microwave plasma synthesis.31 Majority of these synthetic methods involves thermal decomposition of organometallic precursors or metal complexes in the presence of surfactants that enables precise control of both size and its distribution. Despite such advantages, lipophilic nature of the nanoparticle (NP) surfaces and cost of reactants presently make these synthetic methods not viable for biomedical applications.21 Nevertheless, the aqueous coprecipitation of Fe2+ and Fe3+ salts using an alkali remains the most intensively studied, modified and improved method to find economic and environmental friendly pathways for fabrication of controlled metal oxide nanostructures. Most of the industrial applications of magnetite demand highly magnetic nanoparticles with precise knowledge of size, lattice strain/defects and minimal surface passivation. Crystal dimension of nanomaterials affect key colloid properties such as rheology, film gloss, catalytic activity, chemical sensing etc.32 Very often increasing mismatch of magnetic NP size and its saturation magnetization values (and as a result increasing magnetic anisotropy) is reported with decreasing size in magnetite NPs. This is due to the presence of a magnetically dead layer of oxidized 15–30% amorphous component (with a thickness varying between 0.3 and 1.0 nm) over the maghemite shell of magnetite–maghemite core–shell structure.21 We show that there are size-dependent changes in the local structure and oxidation state of the oxide shell, the relative fraction of maghemite increasing at the expense of magnetite as the core dimensions decrease. This size/structure correlation has been explained in terms of morphological and structural disorder arguments.33
In this communication, we report the estimation of the crystal dimensions of magnetite synthesized through surfactant free aqueous coprecipitation route from the TEM, XRD, and magnetization profiles by Langevin fit. The XRD data were analyzed by Rietveld refinement. The magnetite phase was confirmed from the X-ray, TEM, Raman and Fourier transform infrared (FTIR) spectroscopy. The maghemite content in magnetite was determined from the Mössbauer spectral fitting. The magnetic powder was thoroughly characterized by thermal analyses (TG-DTA), surface area analysis, hydrodynamic size and zeta potential measurements. The activation energy for growth of size tailored magnetite crystals was calculated by employing Arrhenius equation. The crystal dimensions were carefully correlated in terms of maghemite layer content over magnetite core in a core–shell model.
Experimental section
Materials
Fe(II) chloride (98%) and Fe(III) chloride (97%) were procured from Sigma Aldrich Chemicals. Ammonia solution (25 wt%) and common solvents ca. acetone, ethanol (analytical grade) etc. were purchased from Merck, India. All the syntheses, washings and dilutions were done with Millipore water (Millipore, specific resistivity 18.2 MΩ cm @ 25 °C). Millipore water for synthetic purposes was purged with XL grade (99.999%) nitrogen gas for deaeration.Experimental
The procedure adopted here is a modification of the method of Shen et al.34 To elucidate the formation process of Fe3O4 nanocrystal in a size tailored manner, 4.2 mmol FeCl2·4H2O and 8.4 mmol FeCl3·6H2O (such that Fe3+/Fe2+ = 2) were dissolved in 35 ml deionized water previously deaerated by purging nitrogen gas in a 100 ml three-neck round bottom flask whose central neck was connected to a water-cooled condenser. A thermometer and a glass pipette for N2 gas purge were connected through thermometer pockets to the other two necks. The reaction mixture was heated slowly to 90 °C over a hot plate magnetic stirrer while continuous stirring. The clear off-yellow color of the suspension turned to muddy orange at ∼80 °C indicating the complete hydrolysis of ferrous and ferric chlorides. After heating the reaction mixture for ∼30 min at 90 °C, ∼7 ml NH3 solution was added while vigorous stirring. The orange colored suspension immediately turned black indicating the formation of magnetite crystals. At this point the net Fe2+ concentration was 100 mM. The temperature was maintained at 90 °C for further 30 min to allow crystal growth. The sequence of magnetite formation from the ferrous and ferric chlorides via hydrated ferrous and ferric oxides during alkali precipitation may be given as35| FeCl3 + 3NH4OH → Fe(OH)3 + 3NH4Cl |
| FeCl2 + 2NH4OH → Fe(OH)2 + 2NH4Cl |
| Fe(OH)2 + 2Fe(OH)3 → Fe3O4 + 4H2O |
The suspension was cooled naturally to ambient temperature. The resultant pH of the reaction mixture was ∼11. The entire experiment was carried out carefully under a closed nitrogen atmosphere. The precipitated black solid was collected by magnetic decantation with a help of permanent neodymium iron boron magnet (520 G strength), and washed five times with 1:1 acetone and methanol mixture. This magnetite is designated as T90. Similarly, magnetites (∼1 g) were produced using the same precursor concentrations at temperatures 33, 45, 60, and 75 °C under identical conditions and were named as T33, T45, T60, and T75. After the final washing, the precipitates were collected and dried at room temperature in a vacuum oven.
The powder XRD patterns were recorded with Bruker D8 Advanced diffractometer equipped with source CuKα1 radiation (λ = 1.5406 Å) with a step size of 0.05° 2θ and a scan speed of 4° min−1. The mean crystallite size and microstrain involved in as-synthesized nano magneto-crystalline samples were estimated by the Cauchy–Cauchy (CC) approach also known as Williamson–Hall36 plot from the linear dependence line-profiling of prominent X-ray diffraction peaks of β2θcosθ versus sinθ as described in eqn (1):
|
β2θcosθ = 2εsinθ + 0.9λ/D
| (1) |
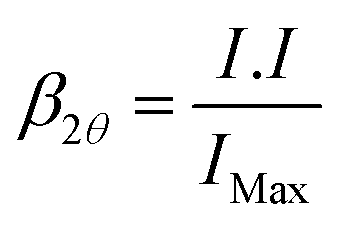 | (2) |
Strain is estimated from the slope (2ε) and the average crystal size (D) from the intercept (0.9λ/D) of the linear regression assuming the particles are spherical. W–H analysis is sometimes used as a complementary method to confirm TEM values and to make them more robust from the statistical point of view. We adopted Rietveld powder structure refinement analysis37 of X-ray powder diffraction step scan data using the JAVA based program MAUD,38 to obtain the structural and microstructural refinement parameters through a least-square method. The experimental profiles are fitted with the most suitable pseudo-Voigt (pV) analytical function37 with asymmetry and the background of each pattern is fitted with a fourth order polynomial function.
Standard Harris analysis was performed on X-ray data of magnetite powders39 to estimate preferred orientation of specific crystal planes and is expressed as texture coefficient C(hikili), following eqn (3),
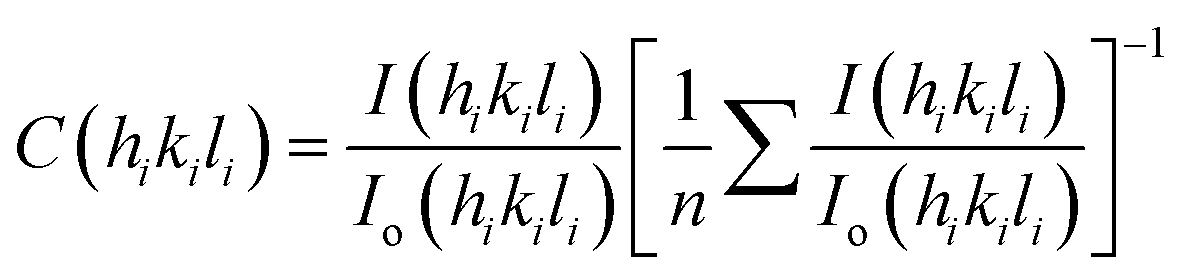 | (3) |
:100 to evaluate the structural aspects of magnetite. The Raman spectrometer is equipped with an optical microscope (Olympus Confocal Raman Optics micrometer), a thermoelectrically cooled (−60 °C) charge-coupled device (CCD) detector and a CCD camera (resolution 1340 × 1100) that can provide a good laser beam. The Raman spectra were collected by 514.5 nm radiation from an argon ion laser (Stellar Pro, 50 mW) on Princeton Instruments Acton SP2500. Samples were compacted into a 1 mm cavity held on an anodized aluminum plate. The laser beam was focused on the sample by a ×50 lens to a spot size of ca. 5–6 μm. Coaxial backscatter geometry was employed for signal collection with spectral resolution of ∼1 cm−1. The Raman shifts were calibrated using the 520 rcm−1 line of a silicon wafer. The spectra were collected using 2.5 mW laser power over the range 1000–200 rcm−1 and accumulation over 10 scans, each with an exposure time of 10 s to reduce noise further. Crystal dimensions can also be estimated from the magnetization curves based on the theory of superparamagnetism as proposed by Bean and co-workers.40–42 To fit the magnetization curves, we assume that individual grains are single crystals without mutual interaction and each particle has an inner single-domain core with the spontaneous magnetization. The magnetization of N number of ideal non-interaction superparamagnetic nanoparticles, each with identical magnetic moment μ, at constant temperature T in magnetic field H is given by Langevin function eqn (4),
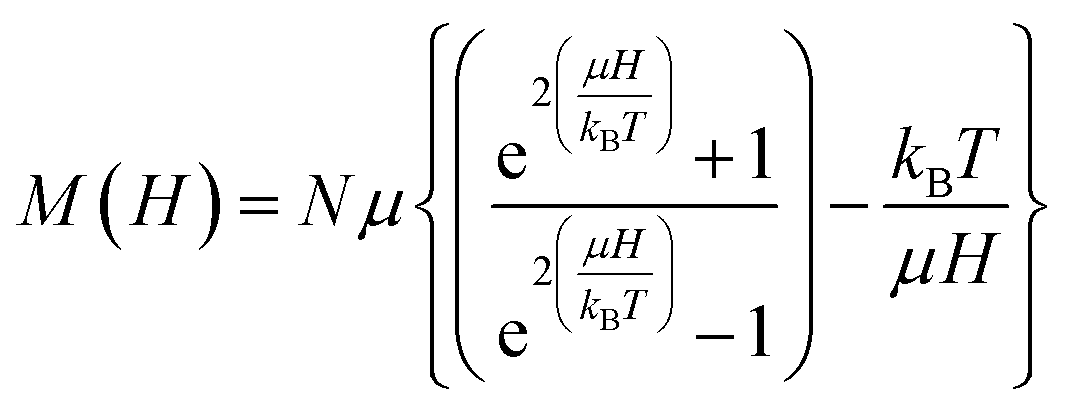 | (4) |
The magnetization curves were fitted using a nonlinear-least squares routine to obtain two parameters: the log-mean single particle moment, μ, and Ms. The size, called “magnetic size”, is significantly smaller than the physical size obtained from TEM. Magnetic measurements of the powder sample were made using VSM (Lakeshore 7305, US) at 298 K and Physical Property Measurement System (PPMS, Cryogen, UK). 57Fe Mössbauer spectra of the two selected specimens T33 and T90 were recorded at room temperature by means of a standard constant acceleration transmission mode with a ∼50 mCi 57Co diffused in rhodium matrix using a α-Fe foil for calibration. The experimental profiles were fitted to Lorentzian functions by least-square method with RECOIL software package. The average size and the morphologies of magnetite nanoparticles and its crystal structure were ascertained by the high resolution transmission electron microscopy (HR-TEM) using a FEI Tecnai 30 G2 S-Twin HR-TEM operated at 300 kV equipped with a Gatan CCD camera. The chemical composition was determined on several crystal grains by using an EDAX spectrometer equipped with high-angle annular dark-field detector with beam scanning capability (Fischione Instruments, Inc., USA) with TIA analysis software. The standard deviation of size was calculated using the formula eqn (5),
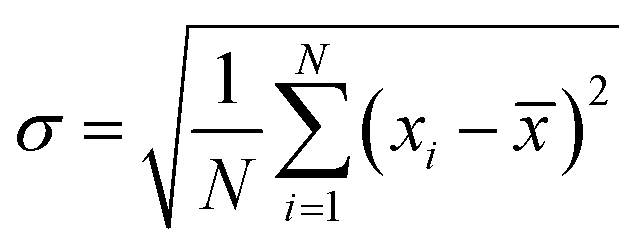 | (5) |
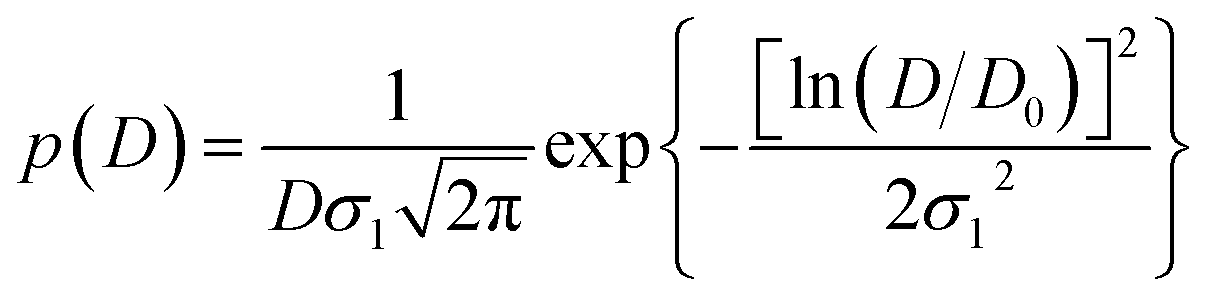 | (6) |
The specific surface area and pore size distribution of the magnetite powders were determined from the N2 adsorption data following BET technique at 77 K using a surface area analyser (Quantachrome Instruments version 10.01). Surface area analyses were conducted on powder samples after degassing them at 200 °C for 3 h. The thermogravimetric (TG) analysis was used to investigate the thermal reactions of the synthesized nanocrystals using a Simultaneous Thermal Analyzer (STA-6000, Perkin-Elmer, The Netherlands) under ultrapure nitrogen purge. The thermograms were collected with a ramp of 10 °C min−1 in the temperature range 50–1000 °C.
Results and discussion
Magnetite nanoparticles were synthesized by the economic, non-toxic aqueous coprecipitation method at different reaction temperatures in the range 33° to 90 °C. The synthesized iron oxide powders (T33 to T90) were typically black in color indicating the presence of magnetite as the dominant phase, although ancillary γ-Fe2O3 may not be completely excluded. The bright field HR-TEM images for selected samples T90, T75, T60 and T33 are shown in Fig. 1 (additional images are provided in Fig. S1 in the ESI†). TEM images show spherical, crystalline and well resolved particles with narrow size distribution in 6–13 nm range. Apparently the particles do not interact with each other at room temperature which will be further investigated by the magnetization measurements in a later section (Fig. 3). The TEM results shown in Fig. 1 reveal that the precipitation temperature increases the average particle size of T33 from 7.76 ± 1.94 nm to 10.84 ± 1.97 nm in T60, while 75 °C reaction temperature further increases the size to 12.32 ± 1.63 nm.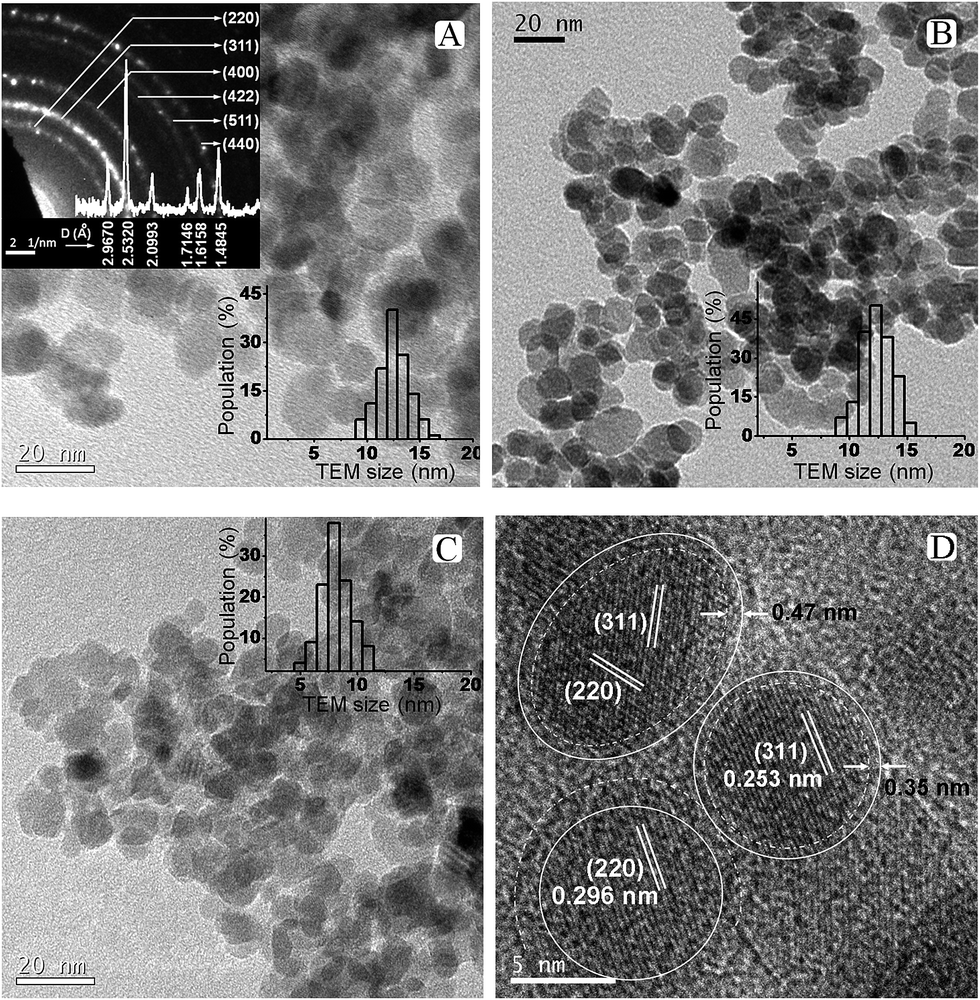 | ||
| Fig. 1 Bright-field TEM images of as synthesized magnetite nanoparticles (A) T90 with its SAED pattern (inset), (B) T75, (C) T33, and (D) HR-TEM of T33 indicating clear crystalline core surrounded by amorphous shell (marked with white line). The size distribution histograms of the corresponding nanocrystals for all the images are presented as inset to each image. | ||
On the other hand, the magnetite synthesized at 90 °C doesn't improve the equilibrium size much but most particles fall in relatively wider 12.42 ± 1.87 nm size range (Fig. 1A). The particle size and number based size distribution of nanoparticles are calculated by probability density function (eqn 6) from the inspection of multiple TEM images on 100–200 particles. These results suggest that the size of the resultant Fe3O4 nanocrystals is strongly dependent on the precipitation temperature. Careful examination of the high resolution images indicates that all the nanoparticles are single crystals and fringes corresponding to predominant (311) and (220) planes could be identified. Selected area diffraction (SAED) patterns of T90 (inset of Fig. 1A) shows clear Debye–Scherrer rings corresponding to (220), (311), (400), (422), (511) and (440) planes with corresponding interplanar spacings 0.2967, 0.2532, 0.2099, 0.1715, 0.1616 and 0.1485 nm respectively (JCPDS card no. 19-0629). The careful observation of HR-TEM images clearly reveals crytallographically different core–shell morphology in nanocrystals of T33 with amorphous shell of thickness ∼0.3–0.5 nm. The atomic ratio of Fe:O of the nanoparticles measured by EDAX (Fig. S1 in ESI†) is 3:4. Within the limits of sensitivity (≤3%), these EDAX data also suggest that the nanoparticles have no detectable impurities: the signal from C and Cu was due to the carbon film over the copper TEM grid.
The X-ray diffraction patterns of Fe3O4 synthesized at different temperatures and Rietveld analysis on the XRD pattern of T33 are shown in Fig. 2. All Bragg's planes could be indexed to face centered cubic inverse spinel magnetite of Fdm, #227 space group with lattice constant a in the range 0.8340–0.8366 nm whereas the same for bulk magnetite is 0.8396 nm (JCPDS card no. 19-0629).
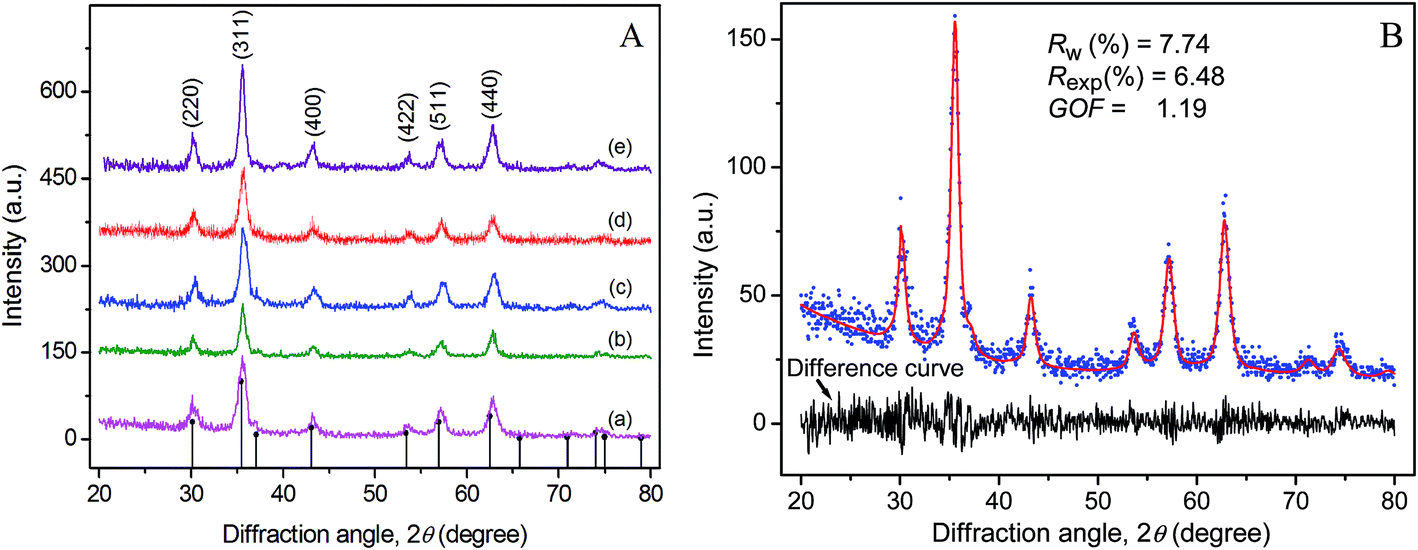 | ||
| Fig. 2 (A) X-ray diffraction patterns of magnetites (a) T33, (b) T45, (c) T60, (d) T75, and (e) T90. The vertical drop lines in (A) are the theoretical Bragg positions for inverse spinel magnetite phase following JCPDS Card no. 19-0629. (B) is a typical Rietveld fit (solid line) for T33 over the corresponding raw XRD data (dots). The noisy layer in (B) is the difference curve between the XRD profile and the Rietveld simulation. | ||
Broad diffraction peaks (Fig. 2A) indicate the nanocrystalline nature of the particles. When the reaction temperature was increased in steps from 33 to 90 °C, a slight progressive narrowing of the X-ray diffraction peaks occurred as a result of crystal growth and reduction of microstrain in the lattice originating from defects. The match of the X-ray pattern with its Rietveld fit and the resultant almost spike less difference pattern indicates the quality of fit in magnetite–maghemite mixed phases in the synthesized sample. The Rietveld analyses of X-ray patterns of T33 and T90 (Fig. S4 in ESI†) allowed us to estimate the relative concentrations of magnetite phase as 89.2 and 87.3% respectively which is compatible with a core–shell model where the shell is 0.2–0.3 nm thick in DTEM = 8.2 and 12.5 nm particles in T33 and T90 respectively. The core–shell structure was also observed from HR-TEM images (Fig. 1). The fit parameters are consistent with reported data in the literature.43 Fig. 3 shows the field dependent magnetic properties of the synthesized Fe3O4 samples (T33 and T90), measured by VSM at room temperature as well as PPMS at different temperatures with higher field resolution. The hysteresis loops apparently pass through the origin which indicates zero coercivity and zero remanence (Fig. 3A), signifying that the samples are in the superparamagnetic state with unstable magnetization at this temperature44 and the samples possess higher magnetization at 250 K due to reduced thermal energy (Fig. 3B).
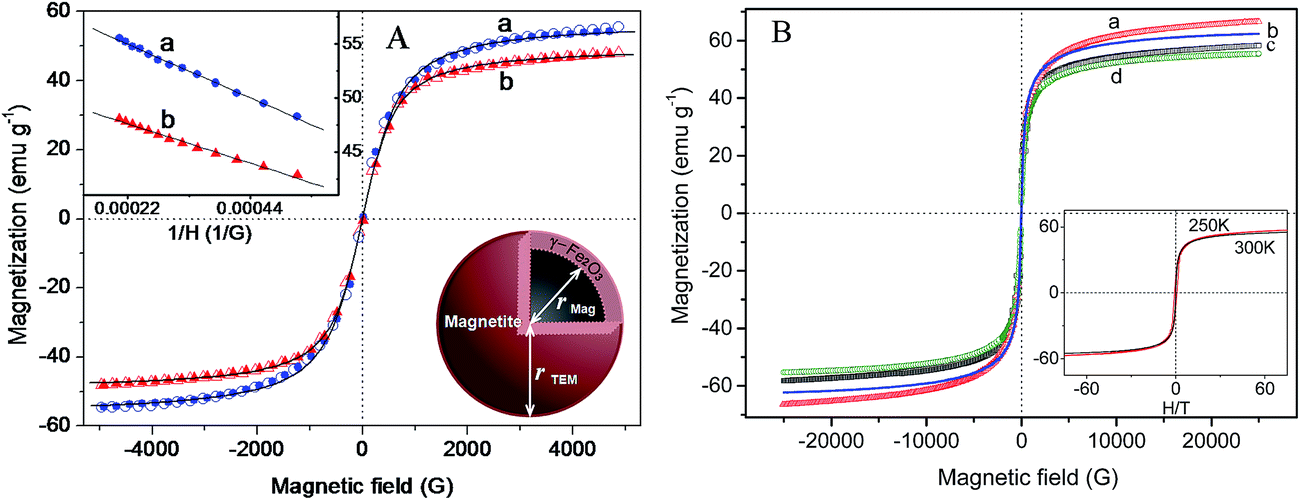 | ||
| Fig. 3 Magnetization versus applied magnetic field data (A) with low field resolution for selected magnetite (a) T33 and (b) T90 specimens and (B) higher field resolution from PPMS at (a) T33 at 250 K, (b) T33 at 300 K, (c) T90 at 250 K and (d) T90 at 300 K and its inset represents the magnetization data as a function of H/T for T90. Typical Langevin fits are represented by solid black lines in (A). | ||
In contrast to saturated magnetization (Ms) of ferromagnetic Fe3O4 nanoparticles, measured by Tian et al.,45 we observed unsaturated magnetic behavior in our synthesized nanoparticles that confirms the superparamagnetic nature. On careful examination of the high resolution magnetization data (Fig. 3B) it is observed that both T33 and T90 display coercivities to the extent of mere 21 and 16 Oe respectively. Appearance of the coercivity may be attributed to the long range magnetic dipolar interactions. The magnetization profiles as a function of H/T (inset of Fig. 3B) are very much close to each other suggesting non-interactive nature of the particles. However, small deviation between the curves in the high field region and the presence of coercive field indicate that the mutual interaction is not absolutely zero i.e., a weak interaction exists among them.46 Due to the asymptotic increase of magnetization for high fields (see Fig. 3), the saturation magnetization value can be obtained from the fitting of the M vs. 1/H curves, extrapolating the magnetization value to 1/H = 0.47 According to inset of Fig. 3A, the observed magnetization for T90 and T33 are 48.44 and 56.11 emu g−1 respectively. The curves were fitted with Langevin function in order to get Ms of samples and the obtained fitting parameters are summarized in Table 1. It is noteworthy to mention that the Ms of our synthesized samples is greater than that of Fe3O4 nanosheets of thickness ∼10 nm and nanorods of length >100 nm and diameter of ∼10 nm,48 but is quite less than bulk Fe3O4 (92 emu g−1)49 and the reduction of Ms may be attributed to the disordered surface spin or spin canting behavior at the surface of the nanoparticles and change in degree of inversion.
| Sample | Exp Ms (emu g−1) | Cal Ms (emu g−1) | μ (emu NP−1) | R2 | Standard error | Dmag (nm) |
|---|---|---|---|---|---|---|
| T33 | 56.11 | 57.22 | 1.48869 × 10−16 | 0.9995 | ±1.04 | 9.86 |
| T90 | 48.44 | 49.86 | 1.71487 × 10−16 | 0.9998 | ±0.54 | 10.83 |
It is worthy to mention that Ms of T33 is higher than that of T90 i.e., bigger particle has lower value of Ms. The magnetic sizes of T90 (Dmag = 10.83 nm) from our computations are smaller than the physical size measured by TEM (DTEM = 12.42 nm). It is reasonable to assume that the difference may be attributed to the presence of magnetically inactive outer layer that is responsible for suppression of magnetization in our synthesized samples.47,50 As it is well known that Fe3O4 having inverse spinel structure in bulk possesses mixed spinel structure in nano-phase and the degree of inversion decreases with increase in particle size. Since T90 has larger size than T33, the reduction of magnetization in case of T90 may be ascribed to the reduction of the degree of inversion. Interestingly, it has been observed that DTEM for T33 (8.2 nm) is smaller than Dmag (9.86 nm) i.e., magnetic size is overestimated by approximately 10% in comparison to TEM size. Such overestimation was previously observed by few researchers and can be explained on the basis of either perturbation in the Langevin function caused by interparticle interactions that was ignored during fitting51 or the consideration of single size distribution of the particles. In order to get a better information, the magnetic sizes (Dmag) were also calculated by using eqn (7),52,53
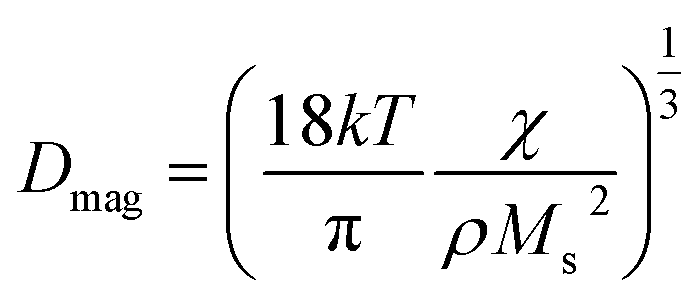 | (7) |
 and density of the material respectively. Eqn (7) gives Dmag as 6.6 nm for T33 and remove the discrepancy that emerged from the Langevin curve analysis.
and density of the material respectively. Eqn (7) gives Dmag as 6.6 nm for T33 and remove the discrepancy that emerged from the Langevin curve analysis.
57Fe Mössbauer spectra of T33 and T90 samples shown in Fig. 4 consist of two sextets and a single doublet pattern depending on the size and crystallinity of the particles. The outer sextet of smaller area corresponds to Fe3+ in tetrahedral (A) sites while the inner sextet with larger area corresponds to Fe3+ and Fe2+ in octahedral (B) sites. However, the weak doublet peak signifies the presence of some particles lying below the critical size for superparamagnetic relaxation at the measurement time-scale. The average hyperfine magnetic field (HMF) decreased in the magnetic powder specimens from T33 synthesized at 33 °C to T90 prepared at 90 °C (Table 2). This drop may be attributed to the lower net magnetic moment per unit formula for T90 as determined by the Fe3+ ion concentration at octahedral site in comparatively larger particles. The relative area ratio of two Fe3+ sextets for both the samples reveals that the concentration of Fe3+ ions at the octahedral (B) site is lower than that at the tetrahedral (A) site.
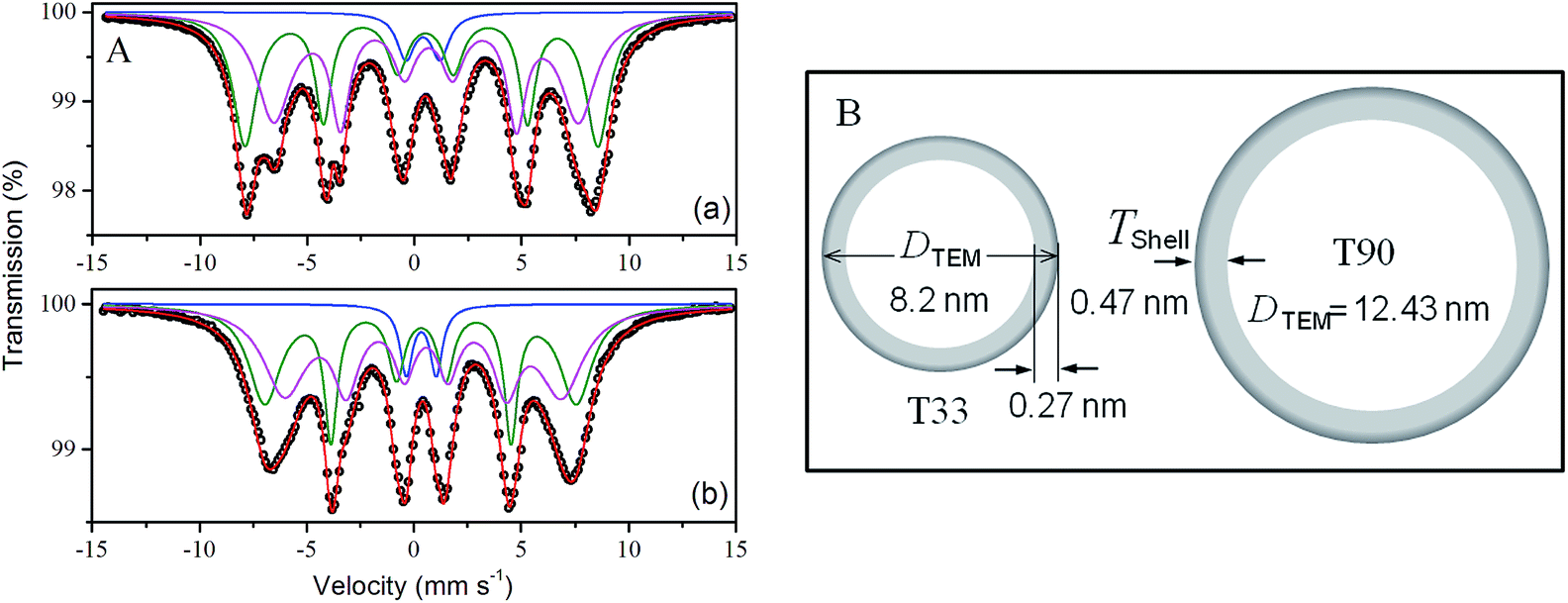 | ||
| Fig. 4 (A) 57Fe transmission Mössbauer spectra of (a) T33 and (b) T90 recorded at room temperature. Symbols represent the experimental data and the continuous lines correspond to the simulated data and (B) schematic representation of core–shell frame-works in T33 and T90. | ||
| Sample | Site | Isomer shift, δa (mm s−1) | Quadrupole splitting, ΔEQa (mm s−1) | Average hyper fine field, Bhfb (Tesla) | Widtha (mm s−1) | Area fractionc (%) |
|---|---|---|---|---|---|---|
| a Standard deviation equal to ±0.03.b Standard deviation equal to ±0.4.c Standard deviation equal to ±0.2. | ||||||
| T33 | Doublet | 0.40 | 1.60 | — | 0.45 | 4.7 |
| Sextet (A) | 0.42 | −0.08 | 51.0 | 0.55 | 42.5 | |
| Sextet [B] | 0.60 | −0.06 | 44.0 | 0.65 | 52.8 | |
| T90 | Doublet | 0.35 | 1.40 | — | 0.35 | 6.2 |
| Sextet (A) | 0.32 | −0.02 | 45.1 | 0.45 | 43.1 | |
| Sextet [B] | 0.52 | −0.08 | 40.0 | 0.60 | 50.7 | |
The isomer shift (δ) values corresponding to tetrahedral (δA) and octahedral (δB) sites of the magnetite specimens (Table 2) illustrate that the s-electron density at the Mössbauer active nuclear site is significantly affected by the rise in precipitation temperature. Nevertheless, the value of the isomer shift of Fe3+ in the A site is higher than that generally reported for the micrometric bulk magnetite,1 indicating a possible charge transfers in the A site also.
Negative quadrupole shift (QS) for both the samples may be attributed to the oblate charge distribution of Fe. The ferric character of the Fe ion is also manifested by the magnitude of the magnetic hyperfine fields and is proportional to the spin of the ferric ion.45 The sextet area leading to a Fetetra3+/Feocta3+,2+ ratio of T33 and T90 is found to be 0.80 (42.5/52.8) and 0.85 respectively, in contrast to the theoretical ratio 0.50. Such intensity ratios, which exceeds 0.50, are called super stoichiometry in oxygen or cationic vacancies.1 In contrast to relative intensity ratio 1:2 corresponding to Fe between A and B sites in bulk inverse spinel magnetite, (Fe3+)A[Fe2+Fe3+]BO4, we observed the global composition of T33 and T90 as,
| T33: (Fe3+)A[Fe0.8142+Fe1.1243+Φ0.062]BO4 |
| T90: (Fe3+)A[Fe0.792+Fe1.143+Φ0.07]BO4 |
tm/τ)1/3. In the measurement of magnetization, the observation time tm ∼ 1 s, and hence the superparamagnetic relaxation can be observed in particles with of size 101 to 102 nm. In a Mössbauer study the measurement time is much shorter (tm ∼ 10−8 s), and therefore manifestation of superparamagnetism is expected in particles of much finer sizes.54
The grain sizes obtained from the W–H plots showed almost linear increase with increasing precipitation temperature indicating crystal growth during precipitation (Fig. S2 in ESI†). A steady increase in the lattice strain was observed (in the range 4.0–7.2 × 10−3) with decrease in the particle size (Fig. S2†).55 It is interesting to note that the estimated (DXRD) from the W–H technique for all the nanocrystals are within DTEM −2.5 nm as the shell of amorphous layer has not contributed to the X-ray diffraction. The deviation is more in the crystals synthesized at higher temperatures. Atoms at the surface and edges are under-coordinated with the presence of broken bonds. As the crystal dimension decreases, the surface area-to-volume ratio increases resulting in a increased broken bond density at the surface causing the remaining bonds to contract spontaneously with an associated increase in bond strength, which in turn produces localized strain.56 A decrease in the lattice parameter to 8.3596 Å was observed when the particle size increased from 7.3 to 8.4 nm as a result of possible higher surface oxidation of magnetite crystals to γ-Fe2O3. The cell constant decreased slightly further to ∼8.3544 Å on increasing size to 9.7 nm and remained almost constant thereafter. W–H analysis of X-ray data confirmed that the lattice parameter and crystal lattice strains in magnetite nanoparticles are primarily dependant on the crystal sizes. The preferential orientation of the crystallites along different crystal planes (hkl), texture coefficients, C(hkl) in the Fe3O4 nanocrystals (Fig. S3†) shows preferentially grown {110} planes to texture coefficient value of 1.323 in T45 because of the growth confinement of (311), (440) and (511) crystal facets and reduced slightly further to 1.288 in T90 (synthesized at 90 °C). Fe3O4 with predominant active (220) planes (T45 to T90) is of great potential in catalytic applications. A C(hkl) value of 1 indicates a particle with randomly oriented crystallites, while a larger value indicates an abundance of crystallites oriented to that (hkl) plane.57 The high-index planes usually have higher surface energy. The energy for different crystal planes is in the order γ(111) < γ(100) < γ(110) < γ(220) for the face-centered-cubic magnetite phase.58
The Arrhenius plot for the samples prepared at temperatures in the range 33–90 °C (Fig. S4†) gave the activation energy from the gradient of the linear regression as 6.15 kJ Mol−1 under the condition of homogeneous growth of nanocrystallites. This energy is responsible for initiating the complex process of nucleation and growth by diffusion as well as secondary growth by Ostwald ripening. In the case of ammonia precipitated magnetite crystals, the activation energy is relatively small as the nanocrystals have large surface area and its poor crystallinity. The growth process involves a dissolution–crystallisation mechanism allowing a decrease of the free enthalpy of the system by reduction of the surface area.42
FTIR spectra on selected magnetite specimens conducted in air are shown in Fig. 5. FTIR spectra also confirm the presence of fcc magnetite in the materials as was previously confirmed from the TEM and XRD data (Fig. 1 and 2). The IR absorption bands in the 630–550 cm−1 range is attributed to the vibrations of Fe–O bonds in tetrahedral and octahedral sites59 and must have been resulted from the split of the ν1 band at ∼570 cm−1.60 The band at ∼440 cm−1 in T90 is due to the octahedral Fe only and corresponds to the ν2 band of Fe–O of bulk magnetite (∼370 cm−1) shifted to a higher wavenumber. The intensity reduction of the band at ∼590 cm−1 from T90 to T33 is probably due to part of the Fe3O4 being reduced to elementary iron.61 The presence of O–H stretching vibration at ∼3411 cm−1 and O–H deformed vibration (bending modes) at 1630 cm−1 are attributed to the presence of coordinated OH groups or water molecules with the unsaturated surface Fe atoms.62 C–O stretching vibrations of CO![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 3 anion at 1400 cm−1 (ν3) in both the samples are due to atmospheric CO2.63 A small absorption band at 2911 cm−1 is due to the νs (–CH) vibrations from the residual solvents left after washing and drying steps. Raman spectra presented in Fig. S6† (see ESI) have also indicated the presence of predominantly magnetite with a small amount of maghemite (γ-Fe2O3) as impurity in the black magnetic products (T75 and T90).
3 anion at 1400 cm−1 (ν3) in both the samples are due to atmospheric CO2.63 A small absorption band at 2911 cm−1 is due to the νs (–CH) vibrations from the residual solvents left after washing and drying steps. Raman spectra presented in Fig. S6† (see ESI) have also indicated the presence of predominantly magnetite with a small amount of maghemite (γ-Fe2O3) as impurity in the black magnetic products (T75 and T90).
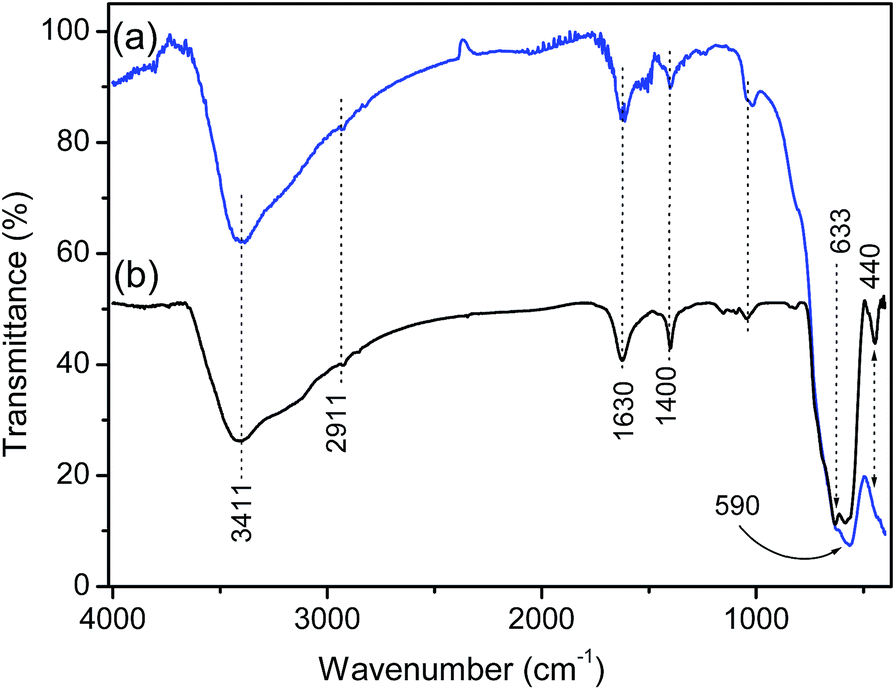 | ||
| Fig. 5 FTIR spectra of the as-obtained (a) T33, and (b) T90 magnetite nanocrystals. | ||
Fig. 6 shows the thermal analysis patterns (TG/DTA), nitrogen adsorption–desorption isotherms and the corresponding pore size distribution profiles of two representative magnetite specimens. The complete analyses of the isotherms are provided in Table 3. Both T33 and T90 showed hysteresis loops of type IV of Brunauer's classification, indicating the presence of mesopores in the powders.
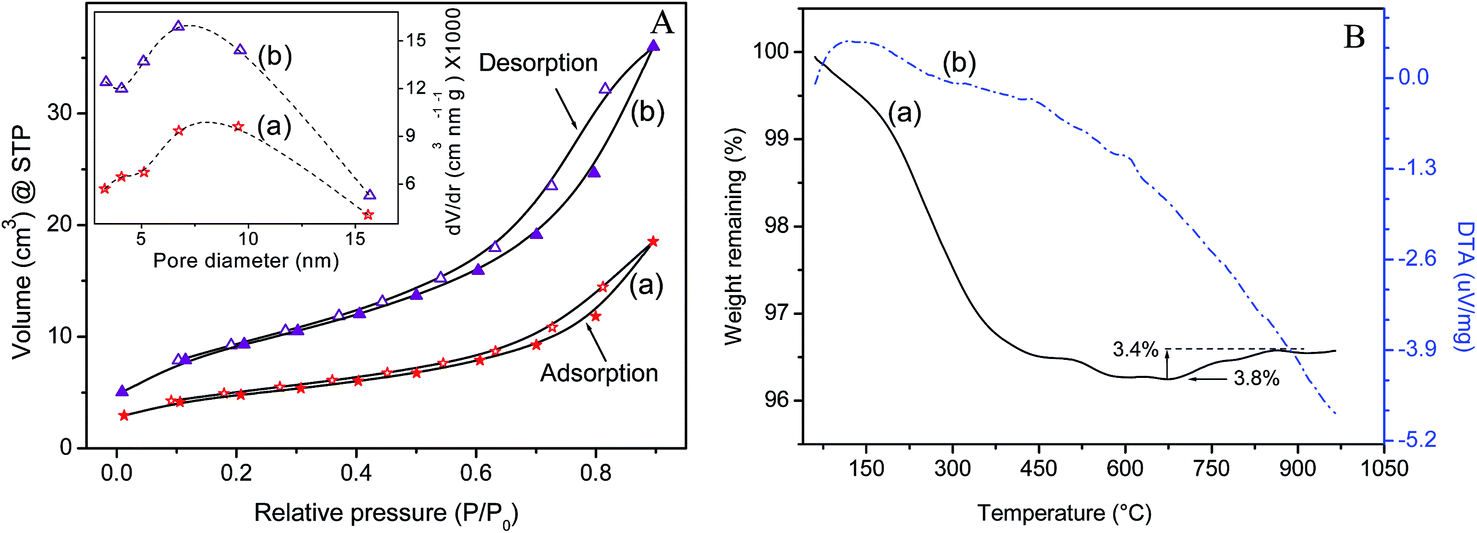 | ||
| Fig. 6 (A) Volume N2 adsorbed versus relative pressure of Fe3O4 nanocrystals at 77 K for the powder samples (a) T90 and (b) T33. Inset shows the plot of pore size distribution derived from the BJH analysis. (B) Thermal analysis curves (a) TG and (b) differential thermal analysis representing the effects of thermal treatment on T90 in pure nitrogen atmosphere. | ||
| Sample | BJH pore distribution desorption | BET surface area (m2 g−1) | Size, DSA (nm) | |||
|---|---|---|---|---|---|---|
| Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) | Total pore volume (cm3 g−1) | |||
| T90 | 51.252 | 0.106 | 9.531 | 0.1197 | 67.974 | 17.04 |
| T75 | 75.917 | 0.145 | 6.782 | 0.1586 | 90.52 | 12.79 |
| T60 | 89.601 | 0.174 | 5.141 | 0.1889 | 105.245 | 11.005 |
| T45 | 85.438 | 0.165 | 6.720 | 0.1836 | 108.474 | 10.68 |
| T33 | 106.230 | 0.195 | 6.729 | 0.2113 | 123.616 | 9.37 |
The plot of dv/dr versus pore size determined by the Barrett–Joyner–Halenda (BJH) method shows that the pores (inset of Fig. 6A) are in the mesoporous range (5–10 nm) and is attributed to the interspaces of the constituent particles. It is obvious that the surface area of magnetite powders decreased with increase in size while increasing the precipitation temperature (Table 3). The particle size (DSA) measured by nitrogen adsorption is somewhat larger than the size estimated from TEM analysis (Fig. 1). This discrepancy can possibly be explained by agglomeration of smaller particles to form larger ones, thereby effectively reducing the collective surface area. The problem of agglomeration in dried NPs was particularly aggravated by the possible magnetic interactions and strong hydrogen bonding among them. The thermogram of magnetite (T90) shows a total weight loss of ∼3.8% in two stages on heating the powder sample to 1000 °C. The first drop in the TG pattern at ∼100 °C is attributed to the physisorbed water remotion. The rate of loss increased till ∼500 °C and it could be attributed to the removal of multi-layers of water of hydration from the surface of the nanoparticles as well as dehydration of iron oxyhydroxide (FeOOH) formed by ambient moisture. A small weight gain of ∼0.4%, observed in the temperature range 685–860 °C is due to oxidation of magnetite to γ-Fe2O3, though the experiment was conducted with continuous nitrogen purge. It is obvious that the thermal behavior of synthetic magnetite depends on the formation temperature, which affects the particle size. The finer magnetite crystals (e.g., 10–20 nm) rapidly undergo transition to maghemite at ∼150 °C; whereas the larger magnetite particles are more thermally stable and do not start the transition to maghemite until about 315 °C.2
The mean hydrodynamic diameter (Z-average) is based on the intensity of scattered photons from the colloidal particles in suspension. The Z-average size measured by dynamic light scattering of T60, is 36.2 nm with a polydispersity index (PDI) of 0.417 (Fig. S5†) though the DTEM for T60 is only 10.84 nm. The PCS size is approximately 3 times larger than the physical size measured by TEM could be due to presence of one-shell hexagonal close-pack clusters of a total of maximum 13 particles with average hydrodynamic diameter of ∼3 × DTEM which is 32.5 nm, close to 36.2 nm. The zeta potential is the electrical potential measured at the shear plane, and represents the portion of the charge that can exert electrostatic attraction and repulsion forces on other particles. The T60 sample exhibit ζ values in the range +44.1 to −43.9 mV for the NP dispersion in the pH range 2.8 to 12. The resulting aqueous dispersions were stable in basic conditions, with the point of zero charge (pHPZC) at pH ∼6.45 which is close to already reported data.64 The corresponding surface reactions may be expressed as
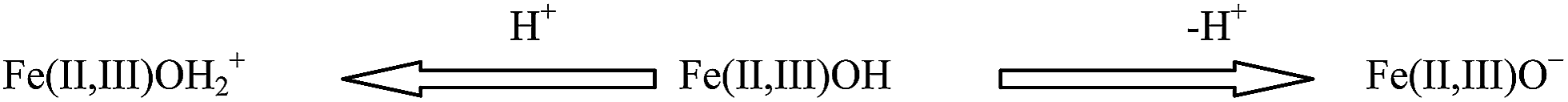
The resulting aqueous dispersions were stable in basic conditions. In acidic pH, the dominating surface species is tentatively Fe(II,III)OH2+, implying positive zeta potentials. With increasing pH, the ζ decreases and Fe(II,III)OH becomes dominating species around pHPZC. At alkaline pH, the surface species Fe(II,III)O− is mainly responsible for the negative ζ.65
Conclusions
Monodisperse magnetite nanoparticles with size in the range 6–13 nm were successfully synthesised by ammonia precipitation technique in the temperature range 33–90 °C. The X-ray diffraction and the Rietveld refinement confirm the inverse spinel structure of cubic phase of magnetite as dominating phase. The activation energy for the growth of nanocrystallites during relatively higher temperature of precipitation was estimated to be ∼6.15 kJ mol−1 which is responsible for initiating the complex process of nucleation and growth by diffusion. W–H technique indicated crystal dimensions within DTEM-2.5 nm as the shell of amorphous layer has not contributed to the X-ray diffraction. The lattice strains calculated were relatively high and in the range 5.5–8.9 × 10−3 with very small variations for samples T33 to T90. The magnetic sizes (Dmag) derived from the Langevin computations were smaller than the physical sizes from TEM analysis due to the supposedly presence of a magnetically “dead” layer of atoms over maghemite shell at the surface. Hence, precipitation method produced monodispersed magnetite nanocrystals with magnetite-maghemite core-shell structures suitable for biological applications.Acknowledgements
The authors are grateful to the Director, Central Glass and Ceramic Research Institute, Kolkata for permission and extending facilities to carry out the above work. SJI and MJ acknowledge CSIR and UGC for their fellowships. Staff members of HR-TEM, XRD, and Central Instrumentation Facility are also acknowledged for their assistance in obtaining data. This work was supported by the 12 FYP CSIR Network project ESC-0103 and CSC-0135.Notes and references
- T. J. Daou, G. Pourroy, S. Begin-Colin, J. M. Greneche, C. Ulhaq-Bouillet, P. Legare, P. Bernhardt, C. Leuvrey and G. Rogez, Chem. Mater., 2006, 18, 4399 CrossRef CAS.
- H. T. Jeng and G. Y. Guo, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 094429 CrossRef.
- H. M. Yang, C. W. Park, M. A. Woo, M. I. Kim, Y. M. Jo, H. G. Park and J. D. Kim, Biomacromolecules, 2010, 11, 2866 CrossRef CAS PubMed.
- Y. Piao, J. Kim, H. Bin Na, D. Kim, J. S. Baek, M. K. Ko, J. H. Lee, M. Shokouhimehr and T. Hyeon, Nat. Mater., 2008, 7, 242 CrossRef CAS PubMed.
- D. K. Kim, M. Mikhaylova, F. H. Wang, J. Kehr, B. Bjelke, Y. Zhang, T. Tsakalakos and M. Muhammed, Chem. Mater., 2003, 15, 4343 CrossRef CAS.
- M. O. Aviles, J. O. Mangual, A. D. Ebner and J. A. Ritter, Int. J. Pharm., 2008, 361, 202 CrossRef CAS PubMed.
- M. Chorny, I. Fishbein, B. B. Yellen, I. S. Alferiev, M. Bakay, S. Ganta, R. Adamo, M. Amiji, G. Friedman and R. J. Levy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8346 CrossRef CAS PubMed.
- S. H. Lee, Z. Y. Lu, S. V. Babu and E. Matijevic, J. Mater. Res., 2002, 17, 2744 CrossRef CAS.
- M. Uchida, M. L. Flenniken, M. Allen, D. A. Willits, B. E. Crowley, S. Brumfield, A. F. Willis, L. Jackiw, M. Jutila, M. J. Young and T. Douglas, J. Am. Chem. Soc., 2006, 128, 16626 CrossRef CAS PubMed.
- K. L. Lai, W. Jiang, J. Z. Tang, Y. Wu, B. He, G. Wang and Z. W. Gu, RSC Adv., 2012, 2, 13007 RSC.
- K. Shimizu, A. Ito, J. K. Lee, T. Yoshida, K. Miwa, H. Ishiguro, Y. Numaguchi, T. Murohara, I. Kodama and H. Honda, Biotechnol. Bioeng., 2007, 96, 803 CrossRef CAS PubMed.
- L. M. Rossi, A. D. Quach and Z. Rosenzweig, Anal. Bioanal. Chem., 2004, 380, 606 CrossRef PubMed.
- M. Shinkai, J. Biosci. Bioeng., 2002, 94, 606 CrossRef CAS.
- C. A. F. Vaz, J. Hoffman, A. B. Posadas and C. H. Ahn, Appl. Phys. Lett., 2009, 94, 022504 CrossRef PubMed.
- H. F. Tian, T. L. Qu, L. B. Luo, J. J. Yang, S. M. Guo, H. Y. Zhang, Y. G. Zhao and J. Q. Li, Appl. Phys. Lett., 2008, 92, 063507 CrossRef PubMed.
- D. Patel, A. Kell, B. Simard, J. X. Deng, B. Xiang, H. Y. Lin, M. Gruwel and G. H. Tian, Biomaterials, 2010, 31, 2866 CrossRef CAS PubMed.
- S. Ghosh, D. Carty, S. P. Clarke, S. A. Corr, R. Tekoriute, Y. K. Gun'ko and D. F. Brougham, Phys. Chem. Chem. Phys., 2010, 12, 14009 RSC.
- S. Ghosh, D. Divya, K. C. Remani and T. S. Sreeremya, J. Nanopart. Res., 2010, 12, 1905 CrossRef CAS.
- U. Colombo, F. Gazzarrini and G. Lanzavecchia, Mater. Sci. Eng., 1967, 2, 125 CrossRef CAS.
- J. Tang, M. Myers, K. A. Bosnick and L. E. Brus, J. Phys. Chem. B, 2003, 107, 7501 CrossRef CAS.
- R. Frison, G. Cernuto, A. Cervellino, O. Zaharko, G. M. Colonna, A. Guagliardi and N. Masciocchi, Chem. Mater., 2013, 25, 4820 CrossRef CAS.
- S. C. Tjong and H. Chen, Mater. Sci. Eng., R, 2004, 45, 1 CrossRef PubMed.
- G. A. Dorofeev, A. N. Streletskii, I. V. Povstugar, A. V. Protasov and E. P. Elsukov, Colloid J., 2012, 74, 675 CrossRef CAS.
- N. J. Tang, W. Zhong, H. Y. Jiang, X. L. Wu, W. Liu and Y. W. Du, J. Magn. Magn. Mater., 2004, 282, 92 CrossRef CAS PubMed.
- H. Wei, N. Insin, J. Lee, H. S. Han, J. M. Cordero, W. H. Liu and M. G. Bawendi, Nano Lett., 2012, 12, 22 CrossRef CAS PubMed.
- Y. F. Wang, Z. W. Zhu, F. Xu and X. L. Wei, J. Nanopart. Res., 2012, 14, 755 CrossRef.
- J. Lee, Y. Lee, J. K. Youn, H. Bin Na, T. Yu, H. Kim, S. M. Lee, Y. M. Koo, J. H. Kwak, H. G. Park, H. N. Chang, M. Hwang, J. G. Park, J. Kim and T. Hyeon, Small, 2008, 4, 143 CrossRef CAS PubMed.
- J. Wan, W. Cai, X. Meng and E. Liu, Chem. Commun., 2007, 5004 RSC.
- J. Pinkas, V. Reichlova, R. Zboril, Z. Moravec, P. Bezdicka and J. Matejkova, Ultrason. Sonochem., 2008, 15, 257 CrossRef CAS PubMed.
- S. Z. Wang, H. W. Xin and Y. T. Qian, Mater. Lett., 1997, 33, 113 CrossRef CAS.
- D. Vollath, D. V. Szabo, R. D. Taylor and J. O. Willis, J. Mater. Res., 1997, 12, 2175 CrossRef.
- O. Masala and R. Seshadri, Annu. Rev. Mater. Res., 2004, 34, 41 CrossRef CAS.
- L. Signorini, L. Pasquini, L. Savini, R. Carboni, F. Boscherini, E. Bonetti, A. Giglia, M. Pedio, N. Mahne and S. Nannarone, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 8 CrossRef.
- L. F. Shen, P. E. Laibinis and T. A. Hatton, Langmuir, 1999, 15, 447 CrossRef CAS.
- I. Nedkov, T. Merodiiska, L. Slavov, R. E. Vandenberghe, Y. Kusano and J. Takada, J. Magn. Magn. Mater., 2006, 300, 358 CrossRef CAS PubMed.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893 CrossRef CAS PubMed.
- H. M. Rietveld, Acta Crystallogr., 1966, 20, 508 CrossRef CAS.
- L. Lutterotti and P. Scardi, J. Appl. Crystallogr., 1990, 23, 246 CrossRef CAS.
- A. Krishnan, T. S. Sreeremya, E. Murray and S. Ghosh, J. Colloid Interface Sci., 2013, 389, 16 CrossRef CAS PubMed.
- C. P. Bean, J. Appl. Phys., 1955, 26, 1381 CrossRef PubMed.
- C. P. Bean and I. S. Jacobs, J. Appl. Phys., 1956, 27, 1448 CrossRef CAS PubMed.
- C. P. Bean and J. D. Livingston, J. Appl. Phys., 1959, 30, S120 CrossRef PubMed.
- V. Yathindranath, L. Rebbouh, D. F. Moore, D. W. Miller, J. van Lierop and T. Hegmann, Adv. Funct. Mater., 2011, 21, 1457 CrossRef CAS.
- Z. C. Xu, C. M. Shen, Y. A. Tian, X. Z. Shi and H. J. Gao, Nanoscale, 2010, 2, 1027 RSC.
- Y. Tian, B. B. Yu, X. Li and K. Li, J. Mater. Chem., 2011, 21, 2476 RSC.
- M. Respaud, J. Appl. Phys., 1999, 86, 556 CrossRef CAS PubMed.
- C. Liu and Z. J. Zhang, Chem. Mater., 2001, 13, 2092 CrossRef CAS.
- H. Shi, L. Tan, Q. Du, X. Chen, L. Li, T. Liu, C. Fu, H. Liu and X. Meng, Dalton Trans., 2014, 43, 12474 RSC.
- S. W. Cao, Y. J. Zhu and J. Chang, New J. Chem., 2008, 32, 1526 RSC.
- R. H. Kodama, J. Magn. Magn. Mater., 1999, 200, 359 CrossRef CAS.
- C. Pascal, J. L. Pascal, F. Favier, M. L. E. Moubtassim and C. Payen, Chem. Mater., 1999, 11, 141 CrossRef CAS.
- B. Y. Yu and S.-Y. Kwak, Dalton Trans., 2011, 40, 9989 RSC.
- T. Ozkaya, M. S. Toprak, A. Baykal, H. Kavas, Y. Koseoglu and B. Aktas, J. Alloys Compd., 2009, 472, 18 CrossRef CAS PubMed.
- N. V. Lukashova, A. G. Savchenko, Y. D. Yagodkin, A. G. Muradova and E. V. Yurtov, Met. Sci. Heat Treat., 2013, 54, 550 CrossRef CAS.
- T. Narayanan, D. Sakthi Kumar, Y. Yoshida and M. Anantharaman, Bull. Mater. Sci., 2008, 31, 759 CrossRef CAS PubMed.
- C. Q. Sun, Nanoscale, 2010, 2, 1930 RSC.
- T. S. Sreeremya, K. M. Thulasi, A. Krishnan and S. Ghosh, Ind. Eng. Chem. Res., 2012, 51, 318 CrossRef CAS.
- C. Yang, J. J. Wu and Y. L. Hou, Chem. Commun., 2011, 47, 5130 RSC.
- A. L. Andrade, D. M. Souza, M. C. Pereira, J. D. Fabris and R. Z. Domingues, Ceramica, 2009, 55, 420 CAS.
- G. Marinescu, L. Patron, D. C. Culita, C. Neagoe, C. I. Lepadatu, I. Balint, L. Bessais and C. B. Cizmas, J. Nanopart. Res., 2006, 8, 1045 CrossRef CAS.
- Y. C. Dong, M. J. Hu, R. G. Ma, H. Cheng, S. L. Yang, Y. Y. Li and J. A. Zapien, CrystEngComm, 2013, 15, 1324 RSC.
- A. Ebrahiminezhad, Y. Ghasemi, S. Rasoul-Amini, J. Barar and S. Davaran, Bull. Korean Chem. Soc., 2012, 33, 3957 CrossRef.
- C. P. Chen, P. Gunawan and R. Xu, J. Mater. Chem., 2011, 21, 1218 RSC.
- J. J. Carlson and S. K. Kawatra, Miner. Process. Extr. Metall. Rev., 2013, 34, 269 CrossRef CAS.
- Z. X. Sun, F. W. Su, W. Forsling and P. O. Samskog, J. Colloid Interface Sci., 1998, 197, 151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11283k |
| This journal is © The Royal Society of Chemistry 2014 |