
Ionic liquid-assisted hydrothermal synthesis of Bi2WO6–reduced graphene oxide composites with enhanced photocatalytic activity†
Hua Lv*,
Yumin Liu,
Jiayuan Hu,
Zijin Li and
Yan Lu
Collaborative Innovation Center of Henan Province for Green Manufacturing of Fine Chemicals, Key Laboratory of Green Chemical Media and Reactions, Ministry of Education, School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, P. R. China. E-mail: hualv2009@163.com; Fax: +86 373 3326336; Tel: +86 373 3326335
First published on 13th November 2014
Abstract
In order to develop highly efficient visible-light-driven photocatalysts, bismuth tungstate (Bi2WO6) was incorporated with reduced graphene oxide (RGO) via an ionic liquid-assisted hydrothermal process. The photocatalytic activities of the as-prepared samples were evaluated by photocatalytic degradation of a rhodamine B aqueous solution under visible light irradiation. Compared with pure Bi2WO6, all the as-prepared RGO–Bi2WO6 composites exhibited significantly enhanced photocatalytic performances for the degradation of rhodamine B. Significantly, the optimum photocatalytic degradation efficiency of RGO–Bi2WO6 composites prepared in the presence of the ionic liquid was about 1.9 times higher than that of Bi2WO6–RGO composites prepared without using the ionic liquid. The excellent photocatalytic performances of RGO–Bi2WO6 composites were correlated with the surface area, light absorption and the separation efficiency of photoinduced electrons and holes. Moreover, the possible mechanism of the enhanced photocatalytic activity for the RGO–Bi2WO6 photocatalyst was proposed on the basis of the experimental results.
1. Introduction
With the increasingly aggravating energy crisis and environmental deterioration, semiconductor photocatalysis, as a green chemical technique, has attracted worldwide attention because of its potential applications in energy conversion and the degradation of organic pollutants in wastewater and the atmosphere.1–3 Among the various semiconductor-based photocatalysts, TiO2 is considered to be one of the most excellent photocatalysts owing to the advantages of nontoxicity, cheapness and chemical stability. However, the band gap of TiO2 is large and it absorbs only small fractions of solar energy. In addition, the practical application of TiO2 photocatalyst is limited by the disadvantage of the moderate photocatalytic activity originating from the rapid recombination rate of photogenerated electron–hole pairs. Considering the utilization of visible light energy and the practical application, it is vital to develop novel photocatalysts with high photocatalytic activities under visible-light irradiation.Bismuth-based photocatalysts, such as Bi2WO6, BiVO4 and BiOBr, might be excited under visible light irradiation and display potential application for water splitting and the degradation of organic pollutants.4,5 Among them, Bi2WO6 with suitable characteristics, such as relatively narrow band gap (Eg = 2.69 eV) and intrinsic layered structure, is regarded as one of the best visible-light-driven photocatalysts.6 However, the slow electron transfer and instability in the process of photocatalytic reaction greatly limit the practical application of Bi2WO6. To overcome the drawbacks, a useful strategy is to prepare composite photocatalyst by doping a certain amount of non-metals such as carbon and nitrogen into Bi2WO6 to improve the electron transfer and suppress the recombination of photogenerated electron–hole pairs effectively. Graphene, as a novel two-dimensional carbon material, has been widely used as an ideal catalyst support material in photocatalytic field owing to its unique planar structure, high specific surface area, superior mobility of charge carriers (>200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 S−1), excellent thermal conductivity (5000 W m−1 K−1), high transparency and chemical stability.7 In 2011, a situ hydrothermal approach was developed for fabrication of graphene oxide–Bi2WO6 composite photocatalyst, which showed an enhance performance for degradation of rhodamine B in comparison with pure Bi2WO6.8 More recently, Zhu and co-workers reported a sonochemical method to fabricate graphene/Bi2WO6 composite, which exhibited improved photocatalytic activity for both H2 and O2 production under visible light irradiation.4
000 cm2 V−1 S−1), excellent thermal conductivity (5000 W m−1 K−1), high transparency and chemical stability.7 In 2011, a situ hydrothermal approach was developed for fabrication of graphene oxide–Bi2WO6 composite photocatalyst, which showed an enhance performance for degradation of rhodamine B in comparison with pure Bi2WO6.8 More recently, Zhu and co-workers reported a sonochemical method to fabricate graphene/Bi2WO6 composite, which exhibited improved photocatalytic activity for both H2 and O2 production under visible light irradiation.4
Room-temperature ionic liquids (ILs), as green and recyclable reaction medium, have attracted increasing attention owing to their unique properties such as good dissolving ability, extremely low volatility, good thermal stability, wide liquid temperature range, high ionic conductivity and wide electrochemical window.5,9 To date, a variety of inorganic nanomaterials with novel morphologies and enhanced properties have been successfully fabricated in ILs, including ring-like ZnO,10 high quality TiO2 nanocrystals,11 BiOBr hollow microspheres,12 nest-like Bi2WO6,13 et al. In these research, ILs play important roles of solvents, templates and reactants for the controlled synthesis of inorganic nanomaterials. If the Bi2WO6/graphene composites can be controlled prepared in the presence of ILs, it can be expected that the combined effects of graphene and ILs would lead to the enhanced photocatalytic activity of the Bi2WO6/graphene composites.
To the best of our knowledge, very little work has been done on the coupling effects of graphene and ILs on the photocatalytic performance of photocatalyst materials. In this work, a simple one-step synthesis of RGO–Bi2WO6 composites was carried out via hydrothermal process in the presence of ionic liquid ([BMIM][BF4]). The performances of the as-prepared products were evaluated by the degradation of Rhodamine B (RhB) under visible light irradiation, showing that the photocatalytic activity of the RGO–Bi2WO6 composites prepared in the present of ILs was higher than that of pure Bi2WO6 or RGO–Bi2WO6 composites prepared without using the ILs. Furthermore, the mechanism of the enhanced photocatalytic activity for RGO–Bi2WO6 composites was also systematically investigated.
2. Experimental section
2.1. Synthesis of RGO–Bi2WO6 nanocomposites
All reagents in this work were of analytical grade and used without further purification. Graphene oxide (GO) was synthesized from natural graphite powder using a modified Hummers method.14 The RGO–Bi2WO6 composites were prepared via an ionic liquid-assisted hydrothermal process in the presence of the GO and hydrazine hydrate. In a typical procedure, 0.291 g of Bi(NO3)3·5H2O was dissolved into 10 mL of 2 M nitric acid solution, while 0.0788 g of (NH4)10W12O41·6H2O was dissolved into a mixture of 10 mL deionized water and 5 mL of [BMIM][BF4]. After the (NH4)10W12O41·6H2O solution was added dropwise into Bi(NO3)3·5H2O solution, the mixture was stirred for 30 min and named as solution A. Simultaneously, a certain amount of GO was dispersed into 15 mL deionized water. The mixture was sonicated for 120 min followed by adding 2 mL of hydrazine hydrate and named as solution B. Subsequently, solution A and solution B were mixed together. The mixture was then transferred into a Teflon-lined autoclave and maintained at 140 °C for 12 h, and cooled to room temperature naturally. Then the as-prepared RGO–Bi2WO6 composites were collected by centrifugation, washed several times with deionized water and absolute alcohol, and finally dried under vacuum at 60 °C for 12 h. In our experiments, the as-prepared RGO–Bi2WO6 composites with 2.5, 5.0, 7.5% wt% RGO were named as 2.5%-RGO–BWO, 5.0%-RGO–BWO and 7.5%-RGO–BWO, respectively. For comparison, pure Bi2WO6 sample was prepared using the same hydrothermal method without the addition of GO and named as BWO. RGO–Bi2WO6 sample with 5.0 wt% RGO was also synthesized without using the ILs and named as 5.0%-RGO–BWO–N.2.2. Characterization
The crystal structures of the samples were determined by XRD (Bruker D8 Advance, Germany) using graphite monochromatic copper radiation (Cu Kα). Morphologies and structures of the samples were observed by SEM (JEOL JSM-63901, Japan) and TEM (JEOL JEM-2100, Japan). The Brunauer–Emmett–Teller (BET) specific surface area was determined, in terms of the N2 adsorption on the power, using a volumetric adsorption apparatus (NOVA Surface Area Analyzer Station A, USA). The optical absorption spectra of samples were recorded by UV-vis diffuse spectrophotometer (Cary5000 UV-vis-NIR) using BaSO4 as a reference. The room temperature photoluminescence (PL) spectra were measured by using a Fluorescence Spectrophotometer (FP-6500, Japan) equipped with a Xenon lamp at an excitation wavelength of 400 nm. The FT-IR spectra were recorded in KBr pellets with Bruker FTIR. Raman spectra were carried out at room temperature by FTS NEXUS with a 514 nm Ar+ laser as an excitation source. XPS analysis was conducted using a PHI Quantera SXM/Auger spectrometer with a monochromated Al Kα X-ray source (1486.6 eV).2.3. Photocatalytic and electrochemical measurements
The photocatalytic performances of the samples were evaluated by photocatalytic degradation of RhB solution under visible light irradiation. A 300 W Xe lamp was used as the light source. All experiments were carried out in a photoreaction apparatus as reported in our previous study.15 In each experiment, 0.1 g photocatalyst was dispersed into 100 mL RhB solution (10 mg L−1). Before illumination, the suspension was sonicated for 10 min and stirred for 30 min in the dark to reach the adsorption–desorption equilibrium. At specific time intervals, about 5 mL of the suspension was withdrawn for further analysis after centrifugation. For the determination of the RhB concentration, UV-vis spectrophotometer (Model T60, Beijing Purkinje General Instrument Company, China) was used at λmax of 553 nm.Electrochemical measurements were performed on a CHI660D electrochemical workstation (Shanghai Chenhua, China) using a standard three-electrode cell with a working electrode, a platinum wire as counter electrode, and a standard Ag/AgCl in saturated KCl as reference electrode. The working electrode was prepared as follows: 50 mg sample was dispersed in 10 mL ethanol and sonicated for 20 min to produce a slurry. The slurry was dip-coated onto a fluorine–tin oxide (FTO) glass electrode with active area of 1.5 cm2. After that, the FTO glass electrode was dried under ambient conditions and then calcined at 400 °C for 1 h. The electrochemical impedance spectra (EIS) were measured at the open circuit potential. The amplitude of the sinusoidal wave was 5 mV, and frequency ranged from 100 kHz to 0.5 Hz.
3. Results and discussion
3.1. Synthesis and characterization
The XRD patterns of GO and RGO–BWO composites with different content of RGO are shown in Fig. 1. The diffraction peak at 9.5° in the XRD pattern of GO corresponds to the layer-to-layer distance (d-spacing) of 0.93 nm. Obviously, the d-spacing of obtained GO is bigger than that of pristine graphite (0.335 nm) attributed to the oxidation and the interlamellar water molecules between the layers.8 Meanwhile, only one peak is observed in the XRD pattern of GO and all other characteristic peaks of graphite cannot be detected, indicating that a highly oxidized GO sample has been obtained.16 The main diffraction peaks of RGO–BWO are similar to that of pure BWO and can well be indexed as orthorhombic symmetry Bi2WO6 crystal phase (JCPDS card no. 39-0256), suggesting that the presence of graphene does not lead to the development of new crystal phase or changes in preferential orientations. Moreover, no obvious diffraction peaks attributed to RGO can be detected in the as-prepared RGO–BWO samples. This phenomenon might be related to the low content, weak diffraction intensity, disruption and well exfoliation of RGO, which is similar with previous reports.17 | ||
| Fig. 1 XRD patterns of GO (a), pure BWO (b), 2.5%-RGO–BWO (c), 5.0%-RGO–BWO (d), 7.5%-RGO–BWO (e), and 5.0%-RGO–BWO–N (f). | ||
The morphology and structure of samples are investigated by SEM and TEM. Fig. 2a displays a typical SEM image of 5.0%-RGO–BWO sample. It clearly shows that large amounts of nest-like structures with an average diameter of 3–5 μm can be obtained by ionic liquid-assisted hydrothermal treatment. The inset of Fig. 2a reveals SEM image of 5.0%-RGO–BWO–N sample synthesized in the absence of ionic liquid. Only a large number of aggregations built by irregular particles appear, suggesting that preparation of RGO–BWO composites with [BMIM][BF4] can readily control the size, morphology and distribution of particles and their interfacial characteristics via their in situ growth and decoration.18 The physical property of [BMIM][BF4] makes it not only act as template and dispersion agents for nanoparticles, but also as active sites for the growth and decoration of nanoparticles on the surfaces of the graphene. From the TEM image (Fig. 2b), the exfoliated GO exhibits flexible sheet-like structure and the characteristic wrinkles can be observed on the edge of GO. As is well known, crumpling and scrolling are intrinsic properties of graphene nanosheets, and the probable reasons are attributed to the thermodynamic stability of the 2D membranes resulting from microscopic crumpling, either by bending or buckling.19 A typical TEM image of 5.0%-RGO–BWO sample (Fig. 2c) illustrates that many small Bi2WO6 particles are uniformly dispersed on the surface and edges of the 2D graphene sheets. As reported previously, Bi2WO6 nanoparticles can interact with graphene by physisorption, electrostatic binding or through charge transfer interactions.8,20 To better understand the hybridization structure between Bi2WO6 and RGO, the 5.0%-RGO–BWO composites were further investigated by high resolution TEM (HR-TEM). The corresponding HR-TEM image (Fig. 2d) shows clear lattice fringes and the spacing of 0.375 nm between the adjacent lattice fringes matches that of (111) crystallographic plane of orthorhombic Bi2WO6. In addition, it can be seen from Fig. 2d that graphene sheet with the basal plane is intimately bridged with Bi2WO6, which can maximally utilize the excellent electron conductivity of graphene and strengthen the electron transport ability of Bi2WO6.
 | ||
| Fig. 2 SEM image of 5.0%-RGO–BWO (inset is the SEM image of 5.0%-RGO–BWO–N) (a); TEM images of GO (b) and 5.0%-RGO–BWO (c and d) at different resolutions. | ||
The FT-IR spectra of GO and 5.0%-RGO–BWO are shown in Fig. S1.† GO shows representative absorptions at 3400 cm−1 (O–H stretching vibration), 1740 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of COOH groups), 1631 cm−1 (aromatic CC vibration), 1406 cm−1 (tertiary C–OH stretching vibration), 1056 cm−1 (C–O stretching vibration), which are in good agreement with previous report.6 For 5.0%-RGO–BWO sample, the main absorption peaks between 500 and 1000 cm−1 can be ascribed to stretching vibration of Bi–O and W–O, and bridging stretching modes of W–O–W.21 In detail, the strong absorption at 730 cm−1 and weak absorption at 580 cm−1 are attributed to the stretching vibration modes of Bi–O and W–O, respectively.22,23 In comparison, the absorption peak of CO (1740 cm−1) decreases in intensity or almost disappears in the FT-IR spectra of 5.0%-GRO–BWO composites, suggesting that the oxygen-containing functional groups in GO is removed during the process of hydrothermal reduction and thus GO is reduced to RGO in the composites.24 Moreover, no peaks corresponding to [BMIM][BF4] are discovered in the FT-IR spectra, revealing the easy removal properties of [BMIM][BF4].
O stretching vibration of COOH groups), 1631 cm−1 (aromatic CC vibration), 1406 cm−1 (tertiary C–OH stretching vibration), 1056 cm−1 (C–O stretching vibration), which are in good agreement with previous report.6 For 5.0%-RGO–BWO sample, the main absorption peaks between 500 and 1000 cm−1 can be ascribed to stretching vibration of Bi–O and W–O, and bridging stretching modes of W–O–W.21 In detail, the strong absorption at 730 cm−1 and weak absorption at 580 cm−1 are attributed to the stretching vibration modes of Bi–O and W–O, respectively.22,23 In comparison, the absorption peak of CO (1740 cm−1) decreases in intensity or almost disappears in the FT-IR spectra of 5.0%-GRO–BWO composites, suggesting that the oxygen-containing functional groups in GO is removed during the process of hydrothermal reduction and thus GO is reduced to RGO in the composites.24 Moreover, no peaks corresponding to [BMIM][BF4] are discovered in the FT-IR spectra, revealing the easy removal properties of [BMIM][BF4].
Raman spectroscopy is a useful nondestructive tool to characterize carbonaceous materials. As shown in Fig. 3, the samples were further characterized by Raman spectroscopy. The Raman spectrum of GO reveals two prominent peaks at around 1582 cm−1 and 1347 cm−1, corresponding to G band (the E2g mode of sp2 carbon atoms) and D band (the breathing mode of the symmetry A1g), respectively. The G and D bands appearing at 1595 cm−1 and 1351 cm−1 for 5.0%-RGO–BWO are similar to those of GO, indicating that the structure of graphene is maintained in the as-prepared composites. Furthermore, it is worth noting that the D/G intensity ratio for RGO in the 5.0%-RGO–BWO sample (0.634) is higher than that of GO (0.411). This change can be ascribed to a decrease in the average size of the sp2 domains upon reduction of exfoliated GO and the creation of a large number of small RGO sheets.19,25
 | ||
| Fig. 3 Raman spectra of GO and 5.0%-RGO–BWO samples. | ||
The chemical composition and element status of the as-prepared samples were further investigated using XPS. Fig. S2† shows the C1s XPS spectra for GO and 5.0%-RGO–BWO composites. In the spectrum of GO (Fig. S2a†), the peak located at 284.5 eV is the characteristic peak of graphitic carbon and corresponds to the C–C bonds, while the deconvoluted peaks at 285.7, 286.8 and 289.2 eV are assigned to C–O, CO, and OC–O functional groups, respectively.26 This result indicates that the as-prepared GO contains a high percentage of oxygen-containing functional groups on its surface. In contrast, the C–O, CO, and OC–O peaks in the spectrum of 5.0%-RGO–BWO composites decrease in intensity, suggesting the deoxygenation of GO, which is in accordance with the FT-IR analysis. The reduction of GO to RGO can significantly enhance the electrical conductivity of the composite, and consequently improve the photocatalytic performance.
The BET surface area and the porous structure of the as-prepared samples were investigated by nitrogen adsorption–desorption measurement. Fig. 6 shows the nitrogen adsorption/desorption isotherms and the corresponding pore-size distribution curves (inset) of the as-prepared samples. As shown in Fig. 4, all isotherms can be categorized as type IV according to the Brunauer–Deming–Deming–Teller (BDDT) classification with distinct hysteresis loops, which is characteristic of porous materials.9 In addition, the hysteresis loops shift approach p/po = 1, indicating the coexistence of mesopores and macropores in the samples.17 The corresponding pore-size distribution curves also confirm that all samples contain non-uniform intra and interaggregated mesopores and macropores with wide pore size distribution. The BET specific surface areas of BWO, 2.5%-RGO–BWO, 5.0%-RGO–BWO, 7.5%-RGO–BWO and 5.0%-RGO–BWO–N samples were calculated to be 5.1, 9.2, 45.5, 16.5 and 12.0 m2 g−1, respectively, indicating that the optimum amount of RGO and the addition of ILs could significantly increase the surface area of the final product. As we know, the average densities of the as-prepared Bi2WO6 with a 0–5.0 wt% graphene loading decrease with the increasing graphene content, leading to the increase of the BET surface areas.17 However, an excessive graphene loading (up to 7.5 wt%) may block the pores of hierarchical Bi2WO6 and consequently decrease the BET specific surface area. Compared with 5.0%-RGO–BWO–N sample, the key factor for higher BET surface area of 5.0%-RGO–BWO composites is attributed to the hierarchical nest-like structure resulting from the addition of ILs.
 | ||
| Fig. 4 N2 adsorption/desorption isotherms and pore size distribution plots (inset) of the samples. | ||
The optical properties of samples were examined using UV-vis diffuse reflectance spectroscopy. As shown in Fig. 5, the pure BWO exhibits its characteristic absorption sharp edge rising at 450 nm, while the RGO–BWO composites show an intense and broad background absorption in the visible light region. Compared with pure BWO, the increased absorption intensity of RGO–BWO composites in the visible light region is attributed to the introduction of blackbody properties of graphite-like materials,27 suggesting that the oxygen-containing functional groups of GO have been well deoxygenated upon the reduction process. In addition, the absorption edges of RGO–BWO composites have obvious redshifts to the long wavelength. This observed redshifts are due to a charge-transfer transition between the RGO and BWO conduction or valance band and is helpful for the efficient utilization of solar energy.28 In addition, the absorbance of the RGO–BWO composites in the visible light region increases with the increasing RGO content in the composites, which could be related to the color of the samples.29 Surprisingly, the 5.0%-RGO–BWO–N sample displays the highest absorption intensity in the visible light region, which might be associated with the difference in morphological structure and further investigation is needed to elucidate this phenomenon.
 | ||
| Fig. 5 UV-vis diffuse reflectance spectra of the as-prepared samples. | ||
Since PL emission mainly results from the recombination of excited electrons and holes, PL spectra is a useful technique to determine the separation efficiency of the photogenerated electron–hole pairs in a semiconductor. Generally, a low PL intensity implies a low recombination rate of the photogenerated electron–hole pairs under light irradiation.30 Fig. 6 displays the PL spectra of the as-prepared samples loaded with different quantities of graphene when the excitation wavelength is 400 nm. It was found that the PL emission spectra of the samples loaded and unloaded RGO exhibited the main peaks at similar positions but with different intensities. All the as-prepared RGO–BWO composites show lower PL intensities in comparison with pure BWO sample, indicating that the introduction of graphene to the composites can accelerate the photogenerated charge carriers transfer, suppress the charge recombination and thus improve the charge separation efficiency.
 | ||
| Fig. 6 Photoluminescence spectra of the as-prepared samples. | ||
To further confirm the separation efficiency of photoinduced electrons and holes, the transient photocurrent responses of pure BWO, 5.0%-RGO–BWO and 5.0%-RGO–BWO–N composites were recorded via on–off cycles of irradiation. As shown in Fig. 7A, the 5.0%-RGO–BWO composites exhibits higher current density, stability, and conversion efficiency than that of pure BWO or 5.0%-RGO–BWO–N composite. It was widely accepted that a higher photocurrent meant the presence of longer living photoinduced electrons and holes pairs and hence the higher the photocatalytic performance would be.31 Thus the separation and transfer of photoinduced electrons and holes are more efficient through coupling graphene and ILs with the photocatalytic material, which is in good agreement with the obtained PL results. On the other hand, EIS measurement was also performed to evaluate differences of the charge transfer resistance and separation efficiency of the photoinduced electron and hole between the photocatalysts. Fig. 7B shows the EIS Nyquist plots of pure BWO, 5.0%-RGO–BWO and 5.0%-RGO–BWO–N composites. The Nyquist plots expanded in the high frequency region are also given in the inset. As shown in Fig. 7B, the arc radius on the EIS Nyquist plot of 5.0%-RGO–BWO was smaller than that of pure BWO or 5.0%-RGO–BWO–N composites, suggesting that a faster interfacial charge transfer and more effective separation of photoinduced electron–hole pairs had occurred.32 This result indicated that the introduction of graphene and ILs into Bi2WO6 during the preparing process can effectively improve the separation efficiency of photoinduced electron–hole pairs, which then could enhance the photocatalytic activity of the composites.
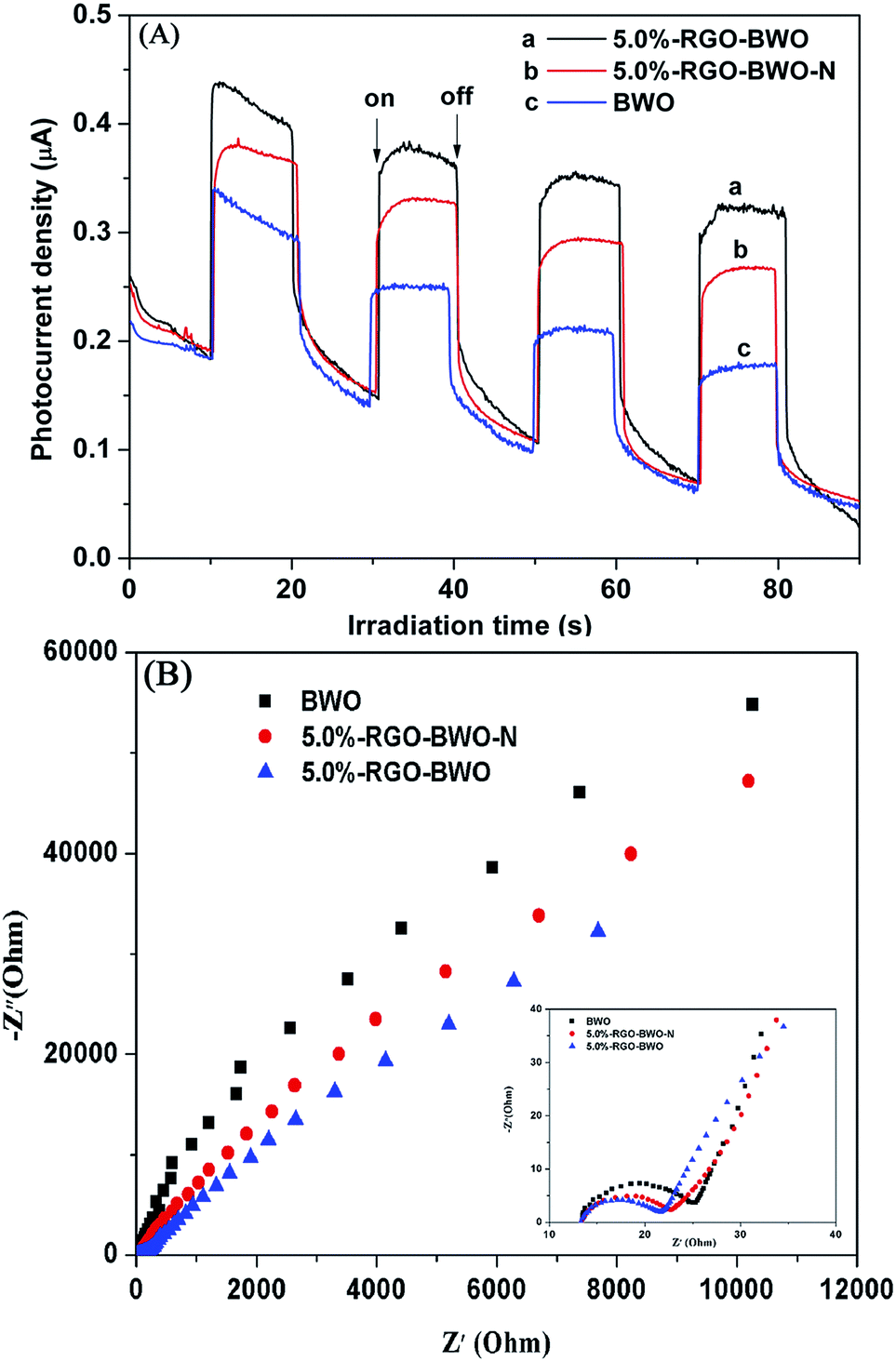 | ||
| Fig. 7 Transient photocurrent response (A) and electrochemical impedance spectroscopy (B) of BWO, 5.0%-RGO–BWO and 5.0%-RGO–BWO–N. | ||
3.2. Enhancement of photocatalytic activity
Rhodamine B (RhB), a widely used dye, was selected as the model pollutant to evaluate the photocatalytic performance of the as-prepared samples. Fig. 8 shows the degradation efficiency of RhB in the presence of different photocatalysts under identical conditions. Ct was the absorption of RhB solution at specific time and C0 is the absorption of the initial RhB solution. A blank test (RhB solution without any photocatalyst) was carried out to estimate the effects of dye self-degradation on the overall photocatalytic performances of Bi2WO6. It can be seen that the degradation of RhB is much lower in the absence of photocatalyst, indicating that the self-degradation of RhB is almost negligible. All RGO modified BWO composites exhibit higher photocatalytic activities in comparison with pure BWO. This is attributed to three factors: (1) the larger BET surface area of RGO modified BWO composites can supply more active adsorption sites and photocatalytic reaction centers, which can lead to an enhanced photocatalytic activity; (2) the presence of RGO in the composites can increase the light absorption intensity and range; (3) RGO can improve the charge separation efficiency. Once graphene is introduced to the Bi2WO6 particles, it can act as an electron collector and transporter to efficiently suppress the recombination of the photogenerated electron–hole pairs, enhance the charge separation efficiency and thus prolong the charge carrier lifetime,33 which has been confirmed from the aforementioned results of PL, photocurrent and EIS spectra of the as-prepared samples. In the composite samples, the photocatalytic activities increase with the increase of RGO content and reach the maximum when the RGO content is 5.0 wt%. With further increase of the RGO content in the composite, the photocatalytic performance deteriorates. This is due to the following reasons: (i) the composites with the RGO content of 5.0 wt% exhibit the highest BET (45.5 m2 g−1), which can offer more surface active sites and make the transfer of charge carrier easier, resulting in the enhanced photocatalytic performance. (ii) An excessive RGO may cause a light harvesting competition between BWO and RGO and thus inhibit the inherent optical absorption of BWO, which results in the decrease of the photocatalytic activities.34 (iii) The excessive RGO can serve as a kind of recombination center instead of providing an electron transport pathway and improve the recombination rate of photoinduced electron–hole pairs in RGO.17,35 Noticeably, the photocatalytic degradation efficiency of 5.0%-RGO–BWO sample (93.1%) is about 1.9 times as high as that of 5.0%-RGO–BWO–N sample (48.8%) after irradiation for 120 min. Compared with 5.0%-RGO–BWO–N composites, the enhanced photocatalytic activity of 5.0%-RGO–BWO composites is mainly ascribed to the nest-like hierarchical structure resulting from the addition of ILs, where the reactant molecules can easily get to the reactive sites on the framework walls of the photocatalysts and thus promotes the photocatalytic degradation.36 Moreover, ILs can act as dispersion agents and make Bi2WO6 nanoparticles uniformly disperse on the surface of the 2D graphene sheets, which is also beneficial for improvement of the charge separation efficiency and photocatalytic activity.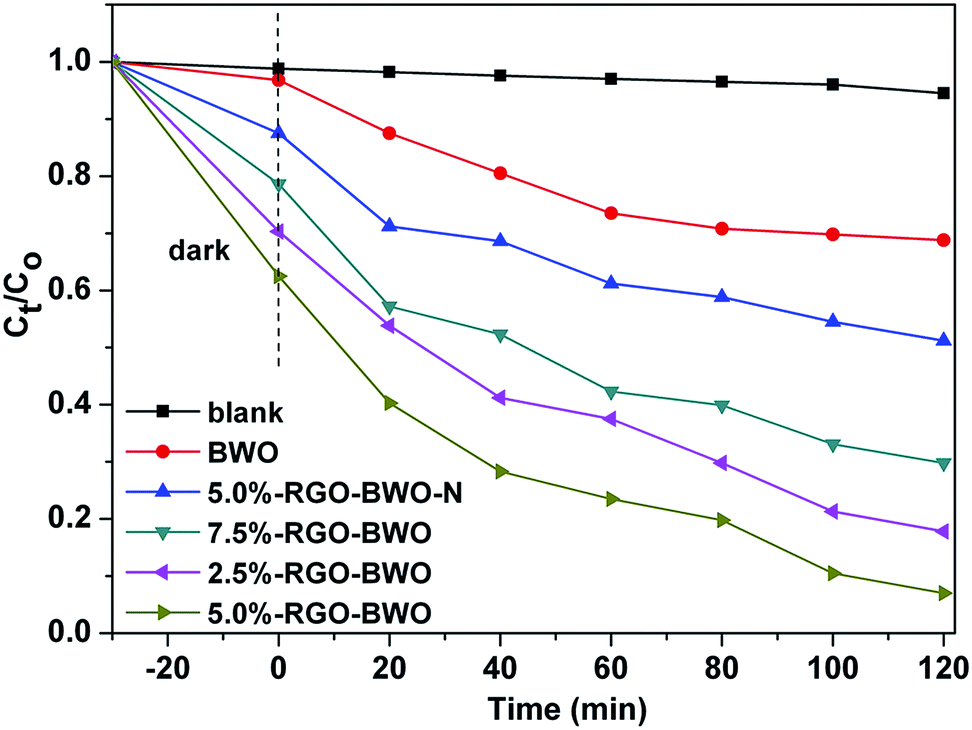 | ||
| Fig. 8 Photocatalytic degradation of RhB under visible light as a function of irradiation time using as-prepared samples. | ||
3.3. Mechanism of the enhanced photocatalytic activity
In order to reveal the possible photocatalytic mechanism involved in RhB degradation over the 5.0%-RGO–BWO composites, trapping experiments have been carried out to determine the main active species during the photocatalytic process (Fig. 9). The photocatalytic activity of 5.0%-RGO–BWO sample exhibits no obvious decrease by the addition of tert-butyl alcohol (TBA, 10 mM, a hydroxyl radicals scavenger), indicating that the hydroxyl radicals ˙OH are not the main oxidative species for 5.0%-RGO–BWO composites. However, once disodium ethylenediaminetetraacetate (EDTA, 10 mM, a photogenerated holes scavenger) or benzoquinone (BQ, 10 mM, a superoxide radicals scavenger) is added to the reaction system, the degradation efficiency of RhB is decreased remarkably. This result means that holes (h+) and superoxide radicals O2˙− should be the main active species in RhB photodegradation and govern the visible light photocatalytic process. To sum up, RGO acts as an acceptor of the generated electrons in RGO–BWO composites and can effectively reduce the photoelectron–hole pair recombination, leaving more hole charge carriers and superoxide radicals O2˙− and thereby improving the photocatalytic activities of samples. In addition, the nest-like hierarchical structure and uniform distribution of BWO resulting from the addition of ionic liquid can enhance the surface area and improve the charge separation efficiency, which also plays an important role in enhancing the degradation of dyes. The mechanism of the improved photocatalytic performance for 5.0%-RGO–BWO composites is summarized and illustrated in Fig. 10. | ||
| Fig. 9 Photocatalytic degradation of RhB by 5.0%-RGO–BWO sample alone and with the addition of EDTA, TAB or BQ. | ||
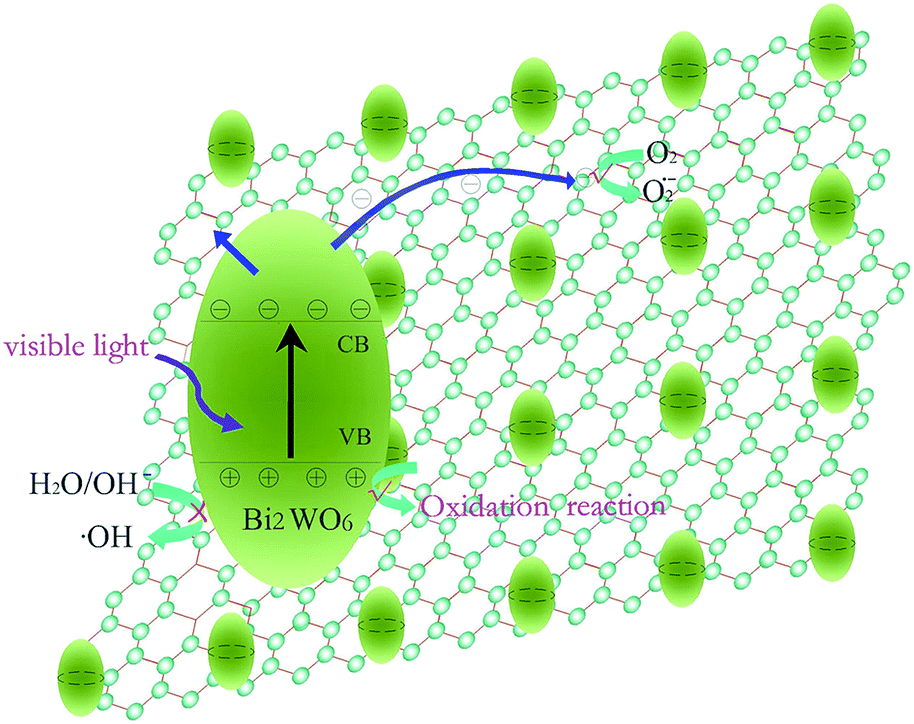 | ||
| Fig. 10 Schematic illustration of RhB degradation over 5.0%-RGO–BWO composites under visible light irradiation. | ||
4. Conclusion
In summary, a series of RGO–BWO composites with various contents of RGO has been successfully synthesized via ionic liquid-assisted one-step hydrothermal route. [BMIM][BF4], an imidazolium-based room-temperature ionic liquid, was used to act as template and dispersion agents, avoiding complicated modification procedures. The photocatalytic activities of all RGO modified BWO composites are significantly enhanced compared to pure BWO. The enhanced photocatalytic performance can be attributed to the combined advantages of the lower charge recombination rate, higher BET surface area and increasing light absorption originating from the introduction of RGO to the composites. Moreover, due to the synergistic contributions of RGO and nest-like Bi2WO6 nanocrystals resulting from the addition of ILs, the RGO/nest-like Bi2WO6 composites with the optimum graphene content of 5.0 wt% show the highest photocatalytic activity in the photocatalytic degradation of RhB dye in comparison with the other as-prepared photocatalysts. Through the utilization of the combined advantages of graphene and ILs, the present work will provide a useful strategy for designing new modified photocatalysts with enhanced activity for environmental purification and other applications.Acknowledgements
The authors gratefully acknowledge the financial support from the National Innovation Experiment Program for University Students (Grant no. 201310476056) and the National Natural Science Foundation of China (Grant no. U1204503).Notes and references
- G. Liu, X. Li, J. Zhao, H. Hidaka and N. Serpone, Environ. Sci. Technol., 2000, 34, 3982–3990 CrossRef CAS.
- Y. Zhang and Y.-J. Xu, RSC Adv., 2014, 4, 2904–2910 RSC.
- Y. Tian, B. Chang, J. Lu, J. Fu, F. Xi and X. Dong, ACS Appl. Mater. Interfaces, 2013, 5, 7079–7085 CAS.
- Z. Sun, J. Guo, S. Zhu, L. Mao, J. Ma and D. Zhang, Nanoscale, 2014, 6, 2186–2193 RSC.
- J. Xia, J. Di, S. Yin, H. Xu, J. Zhang, Y. Xu, L. Xu, H. Li and M. Ji, RSC Adv., 2014, 4, 82–90 RSC.
- Y.-L. Min, K. Zhang, Y.-C. Chen and Y.-G. Zhang, Sep. Purif. Technol., 2012, 86, 98–105 CrossRef CAS PubMed.
- N. Zhang, Y. Zhang and Y. J. Xu, Nanoscale, 2012, 4, 5792–5813 RSC.
- E. Gao, W. Wang, M. Shang and J. Xu, Phys. Chem. Chem. Phys., 2011, 13, 2887–2893 RSC.
- J. Low, J. Yu, Q. Li and B. Cheng, Phys. Chem. Chem. Phys., 2014, 16, 1111–1120 RSC.
- K. Qi, J. Yang, J. Fu, G. Wang, L. Zhu, G. Liu and W. Zheng, CrystEngComm, 2013, 15, 6729–6735 RSC.
- K. Ding, Z. Miao, Z. Liu, Z. Zhang, B. Han, G. An, S. Miao and Y. Xie, J. Am. Chem. Soc., 2007, 129, 6362–6363 CrossRef CAS PubMed.
- J. Xia, S. Yin, H. Li, H. Xu, L. Xu and Y. Xu, Dalton Trans., 2011, 5249–5258 RSC.
- J. Xia, H. Li, Z. Luo, H. Xu, K. Wang, S. Yin and Y. Yan, Mater. Chem. Phys., 2010, 121, 6–9 CrossRef CAS PubMed.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2009, 4, 380–386 CrossRef PubMed.
- Y. Liu, L. Hua and S. Li, Desalination, 2010, 258, 48–53 CrossRef CAS PubMed.
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef.
- H. Ma, J. Shen, M. Shi, X. Lu, Z. Li, Y. Long, N. Li and M. Ye, Appl. Catal., B, 2012, 121–122, 198–205 CrossRef CAS PubMed.
- H. S. Park, B. G. Choi, S. H. Yang, W. H. Shin, J. K. Kang, D. Jung and W. H. Hong, Small, 2009, 5, 1754–1760 CrossRef CAS PubMed.
- J. Shen, M. Shi, B. Yan, H. Ma, N. Li and M. Ye, Nano Res., 2011, 4, 795–806 CrossRef CAS PubMed.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS PubMed.
- D. Ma, S. Huang, W. Chen, S. Hu, F. Shi and K. Fan, J. Phys. Chem. C, 2009, 113, 4369–4374 CAS.
- P. Tang, H. Chen and F. Cao, Mater. Lett., 2012, 68, 171–173 CrossRef CAS PubMed.
- J. Yu, J. Xiong, B. Cheng, Y. Yu and J. Wang, J. Solid State Chem., 2005, 178, 1968–1972 CrossRef CAS PubMed.
- P. Dong, Y. Wang, L. Guo, B. Liu, S. Xin, J. Zhang, Y. Shi, W. Zeng and S. Yin, Nanoscale, 2012, 4, 4641–4649 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- H. Zhang, X. Fan, X. Quan, S. Chen and H. Yu, Environ. Sci. Technol., 2011, 45, 5731–5736 CrossRef CAS PubMed.
- X. Liu, L. Pan, T. Lv, G. Zhu, Z. Sun and C. Sun, Chem. Commun., 2011, 47, 11984–11986 RSC.
- F. B. Li, X. Z. Li, M. F. Hou, K. W. Cheah and W. C. H. Choy, Appl. Catal., A, 2005, 285, 181–189 CrossRef CAS PubMed.
- Z. Qianqian, B. Tang and H. Guoxin, J. Hazard. Mater., 2011, 198, 78–86 CrossRef PubMed.
- D. Wang, G. Xue, Y. Zhen, F. Fu and D. Li, J. Mater. Chem., 2012, 22, 4751–4758 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS.
- E. Gao and W. Wang, Nanoscale, 2013, 5, 11248–11256 RSC.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS PubMed.
- Y.-B. Tang, C.-S. Lee, J. Xu, Z.-T. Liu, Z.-H. Chen, Z. He, Y.-L. Cao, G. Yuan, H. Song and L. Chen, ACS Nano, 2010, 4, 3482–3488 CrossRef CAS PubMed.
- N. Yang, J. Zhai, D. Wang, Y. Chen and L. Jiang, ACS Nano, 2010, 4, 887–894 CrossRef CAS PubMed.
- L. Zhang, W. Wang, Z. Chen, L. Zhou, H. Xu and W. Zhu, J. Mater. Chem., 2007, 17, 2526–2532 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11276h |
| This journal is © The Royal Society of Chemistry 2014 |