
Use of monolithic silicon carbide aerogel as a reusable support for development of regenerable CO2 adsorbent†
Yong Kongab,
Xiaodong Shen*ab,
Sheng Cuiab and
Maohong Fan*cd
aCollege of Materials Science and Engineering, Nanjing Tech University, Nanjing 210009, P. R. China. E-mail: xdshen@njtech.edu.cn; Tel: +86-25-8358-7234
bState Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, Nanjing 210009, P. R. China. E-mail: xdshen@njtech.edu.cn; Tel: +86-25-8358-7234
cDepartment of Chemical and Petroleum Engineering, University of Wyoming, Laramie, WY 82071, USA. E-mail: mfan@uwyo.edu; Tel: +1-307-766-5633
dSchool of Civil and Environmental Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: mfan3@mail.gateche.edu; Tel: +1-404-385-4577
First published on 11th November 2014
Abstract
Monolithic silicon carbide aerogel (MSiCA) was firstly used as a reusable support to develop a novel CO2 adsorbent, i.e. amine functionalized monolithic silicon carbide aerogel (AFMSiCA). Used or exhausted AFMSiCA was separated, treated and re-functionalized by amine to form RF-MSiCA. CO2 capture performance of the resulting adsorbents was investigated in a 1% CO2 flow stream. The results revealed that CO2 adsorption capacities of AFMSiCA at different temperatures ranging from 25 to 75 °C showed little change, indicating that the resulting adsorbent is well-adapted to temperature. The adsorption rate is affected by the combined effect of CO2 adsorption and desorption. Benefiting from the highly unique properties of the aerogel (e.g. high surface area and large pore volume), MSiCA-based adsorbents are dynamic in their CO2 adsorption process. MSiCA could be readily recovered and reused at least 12 times without significant loss of CO2 adsorption performance. The resulting adsorbents presented good stability during cyclic adsorption–regeneration tests. MSiCA is exceptional in practical application for CO2 capture due to its excellent reusability and the regenerability of its amine functionalized counterparts.
Introduction
Concentrations of CO2 in the atmosphere are increasing at an accelerating rate, a phenomenon that has drawn significant attention because of its link to climate change. CO2 capture and sequestration (CCS) is a promising way to control CO2 emissions.1 Among available techniques, amine scrubbing is the current state-of-the-art technology for CO2 capture on an industrial scale. However, this approach shows significant shortcomings, including corrosion, oxidative degradation, high energy consumption for regeneration, and secondary pollution resulting from its high volatility.2 Hence, porous adsorbents functionalized with amines have been extensively studied for CO2 capture because they can overcome the shortcomings of an aqueous amine scrubbing technique.3 Recent investigations have focused on using aerogels as new CO2 capture materials, due to their unique physical properties such as low densities, high surface areas and porosities.4 Moreover, the microstructures and elemental compositions of aerogels can be tailored using solution chemistry via a process known as the sol–gel method.5 Therefore, aerogel-based CO2 adsorbents have been extensively developed recently. Cui et al. achieved a high CO2 adsorption capacity in 10% CO2 mixture stream by using silica aerogel modified with 3-aminopropyltriethoxysilane (APTES).6 Begag et al. developed hydrophobic amine functionalized aerogels by grafting amine groups onto silica backbone and achieved 1.43 mmol g−1 CO2 sorption capacity in simulated flue gas.7 Linneen et al. found that 1 g silica aerogel immobilized with tetraethylenepentamine (TEPA) could adsorb 3.5 mmol CO2 under a dry 10% CO2/Ar stream.8 Wang et al. found that 1 g silica aerogel can adsorb 3.3 mmol CO2 from pure CO2 at 25 °C and 1 atm.9 Masika developed a carbon aerogel for CO2 capture, the CO2 adsorption capacity obtained in pure CO2 at 25 °C and 1 bar was only 2.2 mmol g−1 although the carbon aerogel has a high surface area of 1100 m2 g−1.10 Alhwaige et al. developed chitosan hybrid aerogels for CO2 capture, the CO2 adsorption capacity in pure CO2 at 25 °C is up to 4.15 mmol g−1.11 Lin et al. successfully developed hydrophobic silica aerogel membranes for CO2 capture.12 We have developed a novel aerogel adsorbent based on amine hybrid silica aerogel via facile sol–gel route and supercritical drying, its CO2 adsorption capacity of at 25 °C and 1 atm in a 1% CO2 mixture stream is as high as 5.55 mmol g−1.13 All these reports indicate that aerogels have a promising prospect in CO2 capture.Silicon carbide (SiC) has high hardness, excellent thermal stability and superior chemical inertness,14 therefore, its aerogel counterpart can be promisingly used for CO2 capture. Bhatia et al. developed SiC-derived carbon for CO2 capture.15 However, Monolithic SiC aerogel (MSiCA) has never been used for CO2 adsorbent or support of a CO2 adsorbent. In this work, MSiCA was firstly used as a reusable support to develop a novel CO2 adsorbent, i.e. amine functionalized monolithic silicon carbide aerogel (AFMSiCA). As most of the porous adsorbents for CO2 capture are particulate, they have to be dispersed in sand to prepare a fixed bed to avoid blocking of the reactor when performing CO2 capture process.16 Monolithic aerogels with compact integral framework and porous microstructure can give lower backpressure, higher permeability and better performance compared to particles when they are used in the fixed bed CO2 separation processes.17 Furthermore, researches mostly focus on developing novel adsorbents and improving their CO2 capture performances. Few work pays attention on developing reusable supports or adsorbents, which can reduce the overall CO2 capture costs. The separation and collection of monolithic CO2 adsorbents should be much easier than their particulate counterpart, avoiding secondary pollution and improving utilization efficiency.
Experimental
Preparation of AFMSiCA
Preparation method of MSiCA was reported elsewhere.14a Sol–gel process followed by supercritical drying (SCD) and thermal treatment are used to prepare MSiCA used for the preparation of AFMSiCA. Resorcinol (R), formaldehyde (F, 37% aqueous solution), anhydrous sodium carbonate (C), deionized water (W), and ethanol (EtOH) were mixed at 50 °C with a molar ratio of R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :F:EtOH:W:C = 1:2 : 3.43:2:0.01. The mixture was stirred for 30 min to form RF sol. Tetraethoxysilane (TEOS), EtOH, W and hydrochloric acid (HCl) were mixed at room temperature (25 °C) with TEOS:EtOH:W:HCl prepared at a molar ratio of 1:10.3:2:0.002. The mixture was stirred for 40 min to form silica sol. The RF sol was poured into silica sol with a molar ratio of R:TEOS = 1:2 to form resorcinol-formaldehyde/silica composite (RF/SiO2) sol. The pH value of RF/SiO2 sol was regulated to 9.4 ± 0.1 by ammonium hydroxide. Then RF/SiO2 sol was poured into a polypropylene mould to form RF/SiO2 gel at room temperature (25 °C). RF/SiO2 gel was aged at 60 °C for 24 hours and simultaneously washed with ethanol (3 times, 8 h per wash cycle) to remove water and residual chemicals. The resulting alcogel was dried by SCD to form RF/SiO2 aerogel. RF/SiO2 aerogel was transformed to MSiCA by carbothermal reduction. Thermal treatment of RF/SiO2 aerogel was performed in a tube furnace (72 mm inner diameters of tube). The temperature was first raised to 800 °C with a rate of 2 °C min−1 under argon flow (150 ml min−1), and maintained at that level for 3 h. Subsequently, the flow rate was lowered to 100 ml min−1 and the temperature was raised further to 1500 °C with a rate of 2 °C min−1 and it was maintained at that level for 5 h. At the end of that period, the temperature was lowered to 550 °C, argon was changed to air (200 ml min−1), and excess free carbon was burned off by maintaining the temperature at that level for 3 h.
:F:EtOH:W:C = 1:2 : 3.43:2:0.01. The mixture was stirred for 30 min to form RF sol. Tetraethoxysilane (TEOS), EtOH, W and hydrochloric acid (HCl) were mixed at room temperature (25 °C) with TEOS:EtOH:W:HCl prepared at a molar ratio of 1:10.3:2:0.002. The mixture was stirred for 40 min to form silica sol. The RF sol was poured into silica sol with a molar ratio of R:TEOS = 1:2 to form resorcinol-formaldehyde/silica composite (RF/SiO2) sol. The pH value of RF/SiO2 sol was regulated to 9.4 ± 0.1 by ammonium hydroxide. Then RF/SiO2 sol was poured into a polypropylene mould to form RF/SiO2 gel at room temperature (25 °C). RF/SiO2 gel was aged at 60 °C for 24 hours and simultaneously washed with ethanol (3 times, 8 h per wash cycle) to remove water and residual chemicals. The resulting alcogel was dried by SCD to form RF/SiO2 aerogel. RF/SiO2 aerogel was transformed to MSiCA by carbothermal reduction. Thermal treatment of RF/SiO2 aerogel was performed in a tube furnace (72 mm inner diameters of tube). The temperature was first raised to 800 °C with a rate of 2 °C min−1 under argon flow (150 ml min−1), and maintained at that level for 3 h. Subsequently, the flow rate was lowered to 100 ml min−1 and the temperature was raised further to 1500 °C with a rate of 2 °C min−1 and it was maintained at that level for 5 h. At the end of that period, the temperature was lowered to 550 °C, argon was changed to air (200 ml min−1), and excess free carbon was burned off by maintaining the temperature at that level for 3 h.
To prepare AFMSiCA, MSiCA was immersed in a mixture of water (W), APTES and ethanol (EtOH) at 50 °C for 24 h, the volume ratio of APTES, W and EtOH was prepared at 1:0.2:2. Then MSiCA was taken out from the mixture and dried in an oven at 50 °C to obtain AFMSiCA. To re-functionalize used or exhausted AFMSiCA, AFMSiCA was thermally treated in a furnace at 600 °C for 2 h to burn off the organic moieties, the resulting sample was washed by water under ultrasonic (6 times, 0.5 h per wash cycle) and re-impregnated with APTES via the same procedure for preparing AFMSiCA to obtain re-functionalized MSiCA, denoting as RF-MSiCA-x in which x represents the amine functionalization time. In this paper, x represents 6, 12 or 18.
Sample characterization
Thermogravimetry analysis (TGA) was performed using a Netzsch STA 449C thermogravimetric analyzer under an air flow of 30 ml min−1 at a heating rate of 20 °C min−1. The morphology of the specimens was surveyed by LEO 1530VP scanning electron microscope (SEM) and JEOL JEM-2010 transmission electron microscope (TEM). N2 adsorption–desorption tests were conducted using a Quantachrome Autosorb-iQ analyzer. Samples were treated under vacuum at 90 °C for 3 h before tests. Specific surface areas were calculated using Brunauer–Emmett–Teller (BET) model. Pore-size distributions were obtained using the non-local density functional theory (NLDFT) model. Pore volumes were estimated from the adsorbed amount of N2 at a relative pressure of P/P0 = 0.99. X-ray diffraction (XRD) was performed on a ARL ARLX'TRA X-ray diffractometer with a Cu-Kα radiation to evaluated the phase composition and confirm the formation of SiC nanocrystal.CO2 capture test
The setup for the CO2 capture test at atmospheric pressure is presented in Fig. S1 (ESI†). A fixed bed and a CO2 + N2 mixture gas with a CO2 concentration of 1% were used for CO2 adsorption. Pure N2 was used for desorption through the temperature swing. Adsorbent was placed in the glass tube for CO2 capture. A flow meter (part 9 of Fig. S1†) was used in the apparatus to detect the pressure drop of the fixed bed, i.e. the backpressure of the apparatus. The gas flow rates for adsorption and desorption were calibrated using an Elster American Meter AL17-1 Wet Test Meter and switched to standard state. The adsorption and desorption temperatures were controlled by a MTI GSL-1100X tube furnace. The blank or background of the apparatus was measured when there was no adsorbent in the reactor. All of the calculated capacities in this paper have subtracted the background. Before the CO2 adsorption test, the fresh adsorbents were treated by N2 at 130 °C for 30 min to remove adsorbed moisture and CO2 from the air. For CO2 capture, the temperature of the furnace was stabilized to the temperature for CO2 adsorption. Meanwhile, water vapor (1%) was generated and maintained at a steady state. N2 flow was then switched to the CO2 + N2 mixture gas and water vapor for CO2 adsorption. After finishing CO2 adsorption, the CO2 + N2 mixture gas was changed to N2 to purge the pipes of the apparatus until the outlet CO2 concentration was 0 or very close to 0. The water vapor was cut off, after which the temperature of the furnace was raised to 100 °C for CO2 desorption. For cyclic experiments, the CO2 adsorption process was repeatedly performed on the desorbed adsorbents using the procedure mentioned above. Detail of the calculation method of CO2 adsorption capacity was reported elsewhere.13Results and discussion
It should be noted that APTES moieties in AFMSiCA are physically adsorbed on the surface of SiC nanocrystal or in the pore space of MSiCA. Actually, APTES can be chemically grafted onto the surface of SiC particle. As reported by Williams et al.,18 SiC was first treated in oxygen environment to formation a surface silica layer, the resulting product was exposed to air to obtain moist silanol-terminated SiC, the surface silanol is necessary for the surface graft reaction. The following procedures (reaction, wash and drying) are similar as the surface modification of SBA. The grafted APTES moiety on SiC particle is single layer; hence, the amine loading is low. However, high amine loading should be achieved in order to obtain a desirable CO2 adsorption capacity. Therefore, we used a facile method to deposit APTES onto the surface of SiC particle or into the pore space of SiC network to achieve relatively higher amine loading. It should be also noted that MSiCA is not the adsorbent; it plays an important role of support. AFMSiCA is the CO2 adsorbent.Digital photos of MSiCA, AFMSiCA and RF-MSiCA-x are presented in Fig. S2 (ESI†). All samples remain monolithic shape; revealing that the network of MSiCA is robust and immune to water and high temperature during re-functionalization process. The color of RF-MSiCA-18 is hoary while RF-MSiCA-6 and RF-MSiCA-12 remain the color of MSiCA, which should be due to the oxidation of SiC during thermal treatment and the residual silica particles derived from APTES in the pore space of SiC network, as proved by XRD patterns.
Phase composition of samples
XRD patterns of the samples are obtained, as presented in Fig. S3 (ESI†). The peaks with 2θ values of 35.6°, 41.4°, 60° and 71.8° correspond to the diffraction from the 111, 200, 220 and 311 crystal planes of β-SiC, respectively (PDF#29-1129). A weak peak, which is observed at 2θ = 34.1°, is characteristic of α-SiC (PDF#29-1128).14,19 It can be seen that all the samples remain the characteristic of SiC crystal. No other crystalline phases of silica, carbon or other impurities were obviously detected in the as-prepared MSiCA and AFMSiCA. However, there are amorphous moieties in RF-MSiCA-x, which should derive from the oxidation of SiC framework and residual APTES-based SiO2.Pore structure of the adsorbents
N2 adsorption–desorption isotherms and pore-size distributions of MSiCA, AFMSiCA and RF-MSiCA-x are presented in Fig. 1. Pore structure data of these samples are summarized in Table 1. All samples show type IV isotherms with type H1 hysteresis loops in the IUPAC classification, suggesting that they are typical mesoporous materials with cylindrical pores.19,20 The adsorption of P/P0 < 0.05 attributed to the contribution of micropores (<2 nm) is low, indicating that there is hardly micropores in the adsorbents, which is proved by the pore-size distribution curves. The adsorption from P/P0 = 0.05 to the closed loop is ascribed to the multilayer adsorption on the framework's external surfaces; the abrupt increase of adsorption quantity results from the capillary condensation of N2 in pores. There is no obvious saturation adsorption in the isotherms of MSiCA whereas the saturation adsorption appears in the isotherms of AFMSiCA and RF-MSiCA-x. It reveals that MSiCA has more macropores (>50 nm) than AFMSiCA and RF-MSiCA-x, which can be also proved by pore-size distributions. It can be seen from the pore-size distribution curves that smaller pores disappear and the most probable pores move left from MSiCA to AFMSiCA, due to the blocking of pore space after impregnation.8 Hence, surface area and pore volume present an obvious decrease from MSiCA to AFMSiCA. As observing from the pore-size distribution curves and Table 1, the most probable pores move left and the pore structure data (surface area and pore volume) decrease after amine functionalization and re-functionalization. It is explained by the effect of residual silica particles in the SiC network and the oxidation of SiC network. The residual silica particles which derive from APTES could block the pore space of SiC network. Also, the oxidation of SiC framework results in the decrease of the pore spaces of the adsorbents.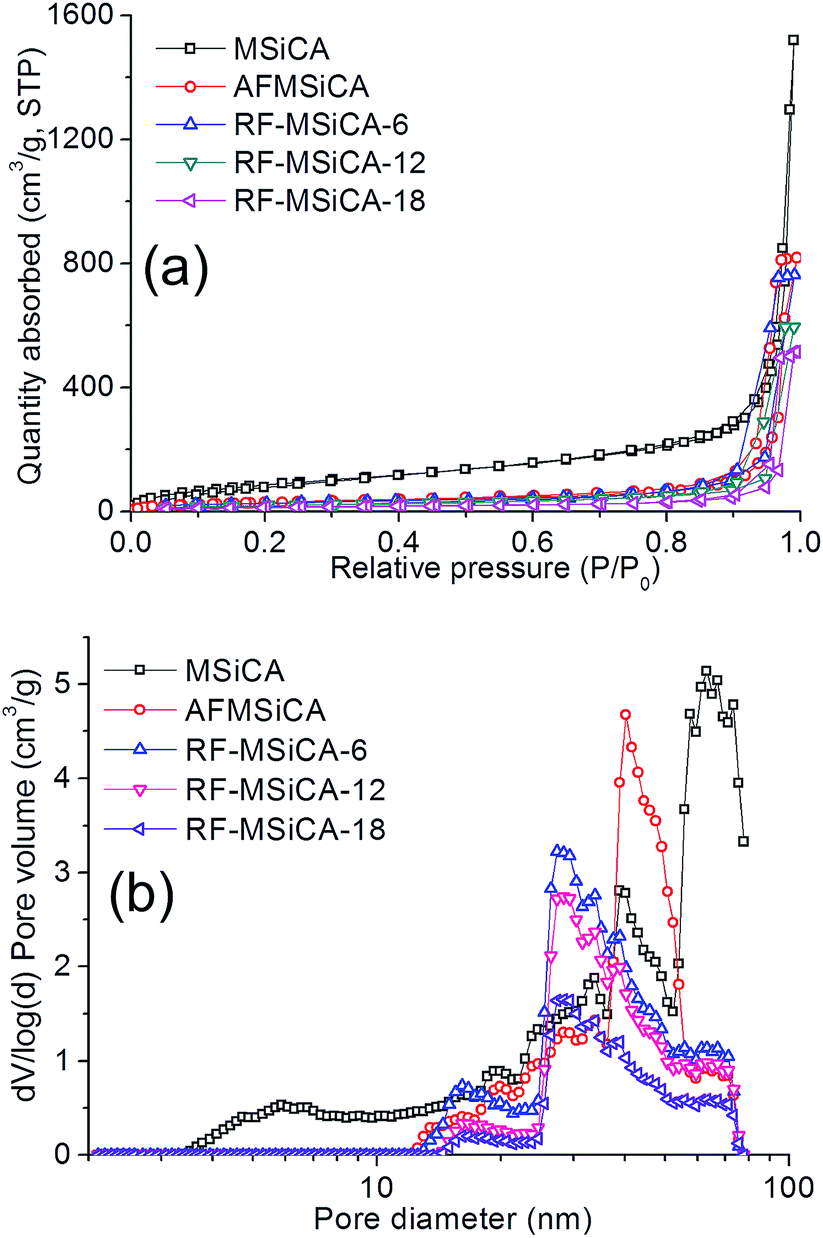 | ||
| Fig. 1 (a) N2 adsorption–desorption isotherms and (b) pore-size distributions of samples. | ||
| Sample | Surface area (m2 g−1) | Pore volume (cm3 g−1) |
|---|---|---|
| MSiCA | 332 | 2.35 |
| AFMSiCA | 108 | 1.27 |
| RF-MSiCA-6 | 99 | 1.18 |
| RF-MSiCA-12 | 72 | 0.92 |
| RF-MSiCA-18 | 43 | 0.56 |
TGA of samples
TG curves of AFMSiCA and RF-MSiCA-x are presented in Fig. 2. TG curve of AFMSiCA suggests that the organic moieties of AFMSiCA can be completely removed after thermal treatment at 550 °C for 2 h. Weight losses of AFMSiCA and RF-MSiCA-x below 155 °C are attributed to moisture and CO2 absorbed from air. Weight losses at 155–250 °C and 250–700 °C result from bonded water and organic moieties, respectively. Increases of weight at high temperature are assigned to the oxidation of SiC. It can be calculated from TG curves that weight loading of organic moieties of APTES in MSiCA is in the range of 38.1–15.7% (Table S1, ESI†). TG curves of RF-MSiCA-x are also measured. The results indicate that the APTES (amine) loading gradually decreases along with the increase of functionalization time. TG curves of MSiCA presented in Fig. S4 (ESI†) reveal that MSiCA is thermally stable in air at under a temperature up to 600 °C.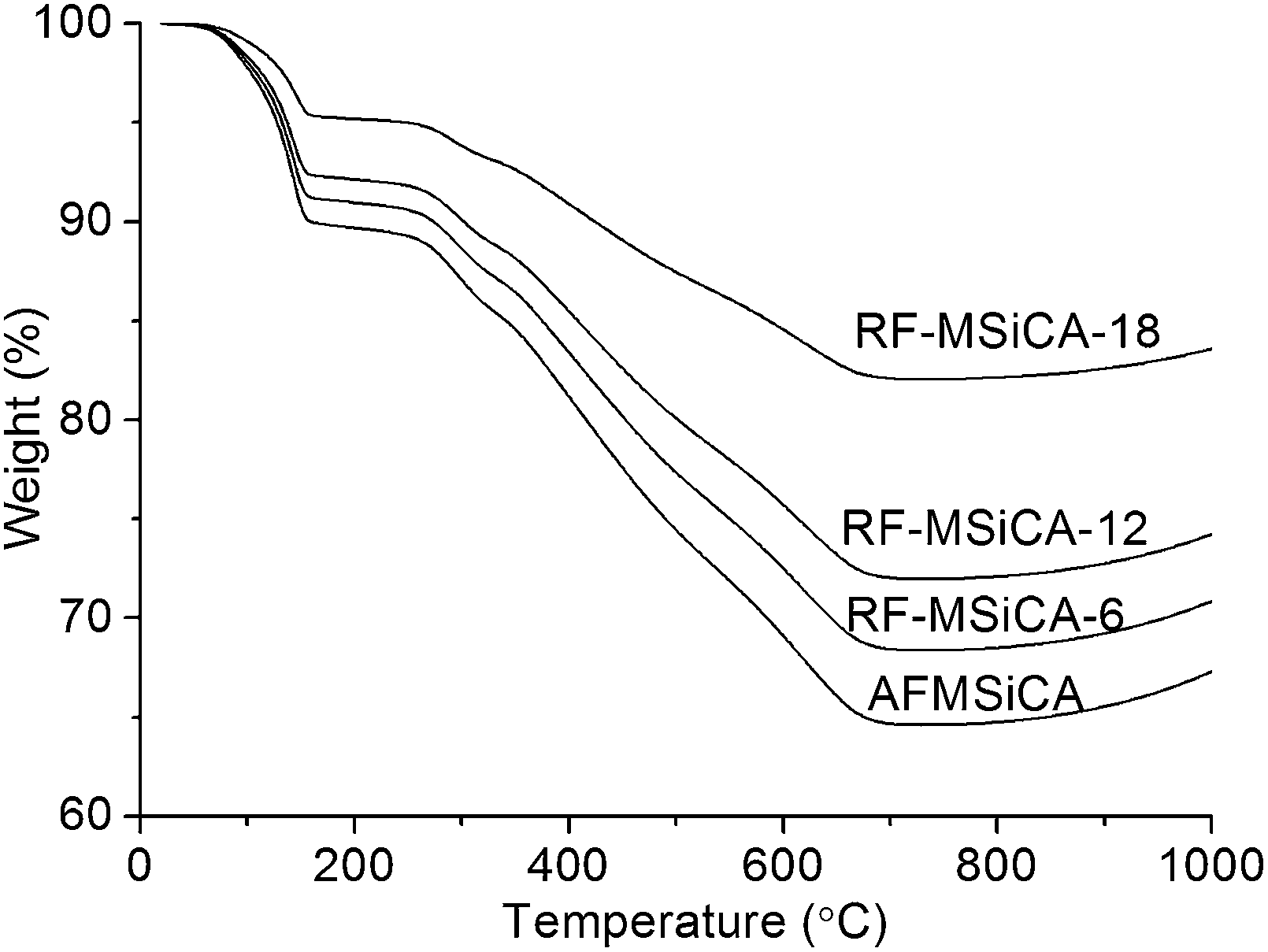 | ||
| Fig. 2 TG curves of AFMSiCA and RF-MSiCA-x. | ||
Morphology of samples
SEM images of MSiCA, AFMSiCA, RF-MSiCA-x are presented in Fig. S5 (ESI†). MSiCA and AFMSiCA present disordered, porous structures of a typical colloidal gel, and nanoparticles are uniformly distributed to form a framework surrounded with irregular pores.21 Porosity gradually decreases along with the increase of functionalization time, which has been proved by the pore volume data presented in Table 1. Meanwhile, particle size increases with the increase of functionalization time. It may be attributed to the effect of residual silica which deposits on the surface of SiC framework and the oxidation of SiC nanocrystal.CO2 capture performance of adsorbents
The CO2 adsorption kinetics and the corresponding CO2 adsorption rates of AFMSiCA at different temperatures in the absence of water are presented in Fig. 3. The CO2 adsorption behavior of AFMSiCA at different temperatures accords with that predicted by thermodynamic theories.22 The total CO2 adsorption capacity of AFMSiCA varies from 1.81, 1.67, 1.53 to 1.00 mmol g−1 when the adsorption temperature increases from 25, 50, 75 to 100 °C. The decrease of capacity from 25 to 75 °C is slow while the decrease of capacity from 75 to 100 °C is sharp. It results from the effect of desorption at 100 °C, indicating that AFMSiCA possesses a desorption temperature around 100 °C. Unlike CO2 adsorption capacity, the CO2 adsorption rate of AFMSiCA at different temperatures seems to follow no rule. The CO2 adsorption rates at 25, 50 and 75 °C are very close, however, the adsorption rate at 100 °C is very different with these obtained at 25–75 °C. The effect of adsorption temperature on the adsorption rate is due to the combined effect of CO2 adsorption and desorption. The CO2 adsorption rate of the adsorbent depends on the competition of these factors. Obviously, higher temperature gives higher CO2 adsorption rate as increasing temperature favors the reaction activity of amine groups. However, the effect of desorption gradually increase along with the increase of temperature. Therefore, adsorption–desorption equilibrium is achieved quickly and the adsorption rate sharply decreases with the adsorption progress when the adsorption temperature increases to 100 °C, indicating that the effect of desorption is dominant in the adsorption–desorption equilibrium at 100 °C. The CO2 adsorption behaviors presented in Fig. 3 suggest that AFMSiCA is strong adaptability to the environment temperature, and therefore suitable for use in various fields and regions. | ||
| Fig. 3 CO2 adsorption kinetics and the corresponding CO2 adsorption rates of AFMSiCA at different temperatures in the absence of water (adsorption time of 20 min; adsorption gas flow rate of 310 ml min−1; weight of adsorbent: 0.145 g): (a) CO2 adsorption kinetics; (b) CO2 adsorption rates. | ||
The effect of water vapor on the CO2 adsorption capacity and the corresponding CO2 adsorption rates of AFMSiCA are presented in Fig. 4. The CO2 adsorption capacities with and without water vapor at 50 °C are 2.12 and 1.67 mmol g−1, respectively. Water is positive to CO2 adsorption capacity and rate in the whole adsorption process. This point has been extensively proved by other amine functionalized adsorbents, and we have also discussed the effect of water on CO2 adsorption performance elsewhere.6,22 It should be noted that the CO2 adsorption capacities obtained from the kinetic curves represent total capacity, while the useful CO2 adsorption capacity in a practical separation process is determined by the breakthrough point (Fig. S6, ESI†).13 The useful CO2 adsorption capacity at 50 °C with and without water are 0.95 and 0.57 mmol g−1, respectively. Therefore, the effective fractions are 44.8% and 34.1%.
 | ||
| Fig. 4 CO2 adsorption kinetics and the corresponding of AFMSiCA at 50 °C with and without water vapour (adsorption time of 20 min; adsorption gas flow rate of 310 ml min−1; weight of adsorbent: 0.145 g; 1% water vapor): (a) CO2 adsorption kinetics; (b) CO2 adsorption rates. | ||
Fig. 5 shows the CO2 adsorption kinetics of AFMSiCA and RF-MSiCA-x at 50 °C. The CO2 adsorption capacity decreases from 2.12 to 1.83, 1.57 and 0.83 mmol g−1 after 6, 12 and 18 times amine functionalization, respectively. It is due to that the content of amine group plays an important role in CO2 adsorption capacity. As presented in Table S1,† APTES loading gradually decreases with the increase of functionalization time, in other words, the surface amine content of the adsorbent decreases along with the increase of the functionalization time. CO2 adsorption reaction of the amine-based adsorbents takes place between the amine group and CO2. Therefore, amine content favors the CO2 adsorption capacity of amine-based adsorbents. The adsorption rate (Fig. S7, ESI†) also presents an obvious decrease with the increase of the functionalization time. It results from the decrease of surface area, pore volume and surface amine content (APTES loading) of the adsorbent after re-functionalization, as presented in Table 1 and S1.† The CO2 adsorption performance of the adsorbents reveals that MSiCA can be effectively recovered and reused as a support of CO2 adsorbent for at least 12 times without fatal decrease of the CO2 adsorption capacity of its amine functionalized counterpart.
 | ||
| Fig. 5 CO2 adsorption kinetics of AFMSiCA and RF-MSiCA-x at 50 °C (adsorption time of 20 min; adsorption gas flow rate of 310 ml min−1; weight of adsorbent: 0.140–0.150 g; 1% water vapor). | ||
The regenerability of CO2 adsorbents is important, as this directly affects their overall CO2 capture costs. Fig. 6 shows the cyclic CO2 adsorption capacities of AFMSiCA and AFMSiCA-12 at 50 °C in the presence of water. The capacities of the adsorbent do not noticeably diminish after 30 cyclic adsorption–desorption tests, decreasing from 2.12 to 2.03 mmol g−1 and from 1.57 to 1.45 mmol g−1 for AFMSiCA and RF-MSiCA-12, respectively. The regenerability of AFMSiCA and RF-MSiCA-12 in humid environment is better than that of amine hybrid silica aerogel and other excellent amine-based CO2 adsorbents, due to their better structure stability with water.13,23 The cyclic stability of the resulting MSiCA-based adsorbents in dry environment is more excellent, as presented in Fig. S8 (ESI†). This encouraging result shows that the adsorbent based on MSiCA is exceptional in regenerability.
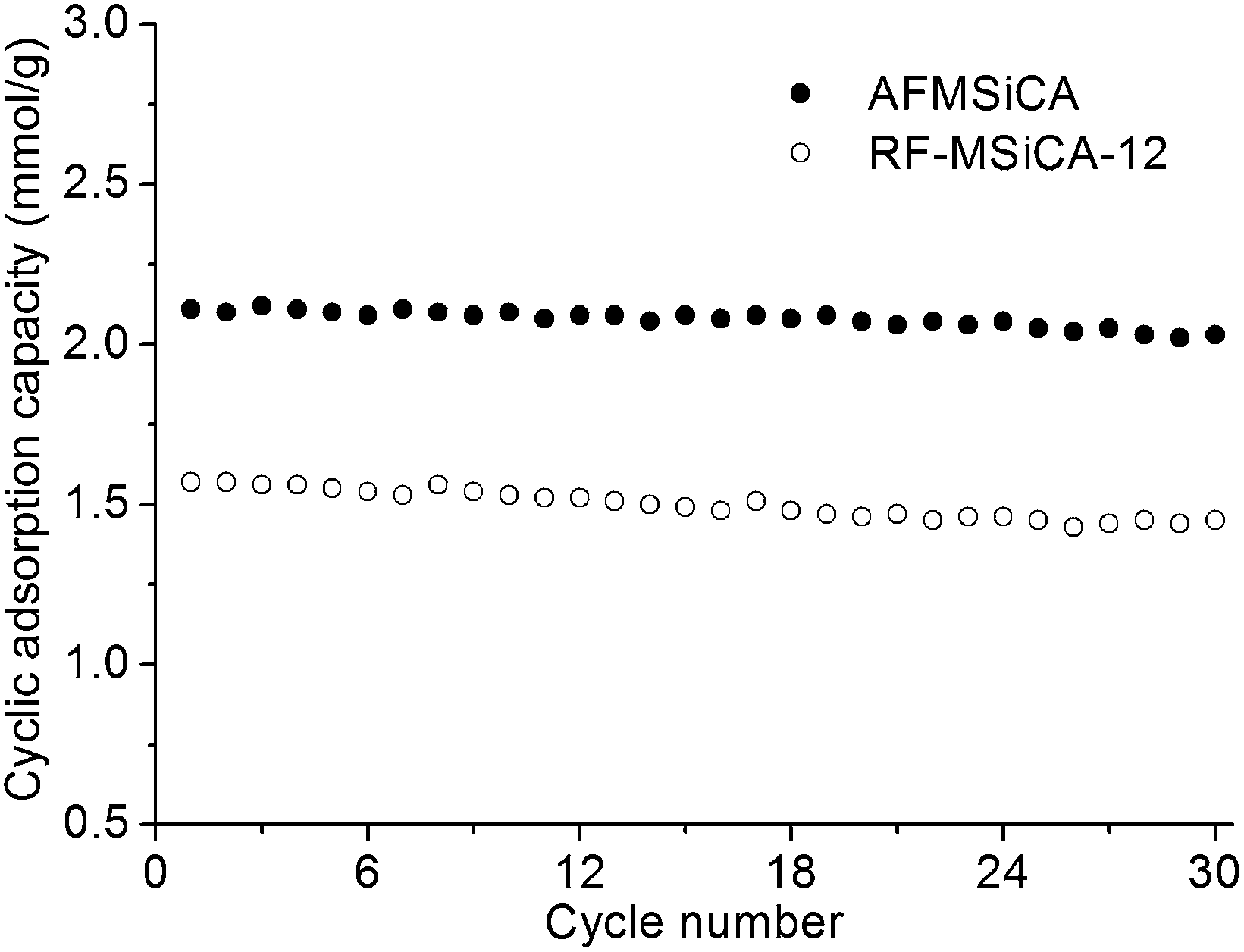 | ||
| Fig. 6 Cyclic adsorption capacities of AFMSiCA and RF-MSiCA-12 at 50 °C (adsorption time of 20 min; adsorption gas flow rate of 310 ml min−1; weight of adsorbent: 0.150 g; 1% water vapor; desorption gas flow rate of 310 ml min−1; desorption temperature of 100 °C). | ||
Conclusions
MSiCA with high surface area and large pore volume is a robust and effective support for preparing amine functionalized CO2 adsorbent. AFMSiCA was prepared by wet impregnation of MSiCA with APTES. Used or exhausted AFMSiCA was separated, treated and re-functionalized by APTES to reactivate and regenerate its CO2 capture ability. MSiCA could be readily recovered and reused at least for 12 times without significant loss of CO2 adsorption performance. The resulting MSiCA-based adsorbents are dynamic in CO2 capture process and present good stability during cyclic adsorption–regeneration tests with and without water. The resulting adsorbent is strong adaptability to the environment temperature, and therefore suitable for use in various fields and regions. MSiCA is exceptional in practical application due to its excellent reusability and the regenerability of its amine functionalized counterparts.Acknowledgements
The authors acknowledge the supports from Jiangsu Planned Projects for Postdoctoral Research Funds (1402016A), the Priority Academic Program Development of Jiangsu Higher Education Institution (PAPD) and the Program for Changjiang Scholars and Innovative Research Team in University (no. IRT1146).Notes and references
- (a) X. P. Zhang, X. C. Zhang, H. F. Dong, Z. J. Zhao, S. J. Zhang and Y. Huang, Energy Environ. Sci., 2012, 5, 6668–6681 RSC; (b) J. M. Zhang, J. Sun, X. C. Zhang, Y. S. Zhao and S. J. Zhang, Greenhouse Gases: Sci. Technol., 2011, 1, 142–159 CrossRef CAS; (c) W. M. Budzianowski, Energy, 2012, 41, 280–297 CrossRef CAS PubMed; (d) D. F. Guo, H. Thee, C. Y. Tan, J. Chen, W. Y. Fei, S. Kentish, G. W. Stevens and G. da Silva, Energy Fuels, 2013, 27, 3898–3904 CrossRef CAS; (e) R. Li, X. Q. Ren, X. Feng, X. G. Li, C. W. Hu and B. Wang, Chem. Commun., 2014, 50, 6894–6897 RSC.
- (a) S. S. Feng, W. Li, Q. Shi, Y. H. Li, J. C. Chen, Y. Ling, A. M. Asiri and D. Y. Zhao, Chem. Commun., 2014, 50, 329–331 RSC; (b) X. Q. Fan, L. X. Zhang, G. B. Zhang, Z. Shu and J. L. Shi, Carbon, 2013, 61, 423–430 CrossRef CAS PubMed; (c) F. S. Li, W. Qiu, R. P. Lively, J. S. Lee, A. A. Rownaghi and W. J. Koros, ChemSusChem, 2013, 6, 1216–1223 CrossRef CAS PubMed.
- (a) B. T. Zhang, M. Fan and A. E. Bland, Energy Fuels, 2011, 25, 1919–1925 CrossRef CAS; (b) L. Ma, R. Bai, G. Hu, R. Chen, X. Hu, W. Dai, H. F. Dacosta and M. Fan, Energy Fuels, 2013, 27, 5433–5439 CAS; (c) B. Dutcher, M. Fan, B. Leonard, M. D. Dyar, J. Tang, E. A. Speicher, P. Liu and Y. Zhang, J. Phys. Chem. C, 2011, 115, 15532–15544 CrossRef CAS; (d) B. Dutcher, M. H. Fan, S. Cui, X. D. Shen, Y. Kong, A. G. Russell, P. McCurdy and M. Giotto, Int. J. Greenhouse Gas Control, 2013, 18, 51–56 CrossRef CAS PubMed; (e) N. A. Brunelli, S. A. Didas, K. Venkatasubbaiah and C. W. Jones, J. Am. Chem. Soc., 2012, 134, 13950–13953 CrossRef CAS PubMed; (f) F. Rezaei, R. P. Lively, Y. Labreche, G. Chen, Y. F. Fan, W. J. Koros and C. W. Jones, ACS Appl. Mater. Interfaces, 2013, 5, 3921–3931 CrossRef CAS PubMed.
- (a) M. Schwan and L. Ratke, J. Mater. Chem. A, 2013, 1, 13462–13468 RSC; (b) A. Fidalgo, J. P. S. Farinha, J. M. G. Martinho and L. M. Ilharco, J. Mater. Chem. A, 2013, 1, 12044–12052 RSC; (c) J. Guo, B. N. Nguyen, L. C. Li, M. A. B. Meador, D. A. Scheiman and M. Cakmak, J. Mater. Chem. A, 2013, 1, 7211–7221 RSC; (d) J. H. Li, J. Y. Li, H. Meng, S. Y. Xie, B. W. Zhang, L. F. Li, H. J. Ma, J. Y. Zhang and M. Yu, J. Mater. Chem. A, 2014, 2, 2934–2941 RSC.
- (a) S. Han, J. Z. Wang, S. Li, D. Q. Wu and X. L. Feng, J. Mater. Chem. A, 2014, 2, 6174–6179 RSC; (b) Y. F. Lin and C. Y. Chang, RSC Adv., 2014, 4, 28628–28631 RSC; (c) A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243–4265 CrossRef CAS PubMed; (d) Y. Kong, Y. Zhong, X. D. Shen, S. Cui and M. Fan, Microporous Mesoporous Mater., 2014, 197, 77–82 CrossRef CAS PubMed.
- S. Cui, W. Cheng, X. D. Shen, M. Fan, A. T. Russell, Z. Wu and X. Yi, Energy Environ. Sci., 2011, 4, 2070–2074 CAS.
- R. Begag, H. Krutka, W. Dong, D. Mihalcik, W. Rhine, G. Gould, J. Baldic and P. Nahass, Greenhouse Gases: Sci. Technol., 2013, 3, 30–39 CrossRef CAS.
- (a) N. Linneen, R. Pfeffer and Y. Lin, Microporous Mesoporous Mater., 2013, 176, 123–131 CrossRef CAS PubMed; (b) N. N. Linneen, R. Pfeffer and Y. Lin, Ind. Eng. Chem. Res., 2013, 52, 14671–14679 CrossRef CAS.
- Z. Wang, Z. Dai, J. Wu, N. Zhao and J. Xu, Adv. Mater., 2013, 25, 4494–4497 CrossRef CAS PubMed.
- E. Masika and R. Mokaya, RSC Adv., 2013, 3, 17677–17681 RSC.
- A. A. Alhwaige, T. Agag, H. Ishida and S. Qutubuddin, RSC Adv., 2013, 3, 16011–16020 RSC.
- (a) Y. F. Lin, C. C. Ko, C. H. Chen, K. L. Tung and K. S. Chang, RSC Adv., 2014, 4, 1456–1459 RSC; (b) Y. F. Lin, C. H. Chen, K. L. Tung, T. Y. Wei, S. Y. Lu and K. S. Chang, ChemSusChem, 2013, 6, 437–442 CrossRef CAS PubMed.
- (a) Y. Kong, G. Jiang, M. Fan, X. D. Shen, S. Cui and A. G. Russell, Chem. Commun., 2014, 50, 12158–12161 RSC; (b) Y. Kong, G. Jiang, M. Fan, X. D. Shen and S. Cui, RSC Adv., 2014, 4, 43448–43453 RSC.
- (a) Y. Kong, X. D. Shen, S. Cui and M. H. Fan, Ceram. Int., 2014, 40, 8265–8271 CrossRef CAS PubMed; (b) Y. Kong, Y. Zhong, X. D. Shen, L. H. Gu, S. Cui and M. Yang, Mater. Lett., 2013, 99, 108–110 CrossRef CAS PubMed.
- S. K. Bhatia and T. X. Nguyen, Ind. Eng. Chem. Res., 2011, 50, 10380–10383 CrossRef CAS.
- (a) G. Qi, Y. Wang, L. Estevez, X. Duan, N. Anako, A. –H. A. Park, W. Li, C. W. Jones and E. P. Giannelis, Energy Environ. Sci., 2011, 4, 444–452 RSC; (b) J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS PubMed; (c) J. H. Drese, S. Choi, R. P. Lively, W. J. Koros, D. J. Fauth, M. L. Gray and C. W. Jones, Adv. Funct. Mater., 2009, 19, 3821–3832 CrossRef CAS.
- B. Gawel, K. Gawel and G. Oye, Materials, 2010, 3, 2815–2833 CrossRef CAS PubMed.
- E. H. Williams, A. V. Davydov, A. Motayed, S. G. Sundaresan, P. Bocchini, L. J. Richter, G. Stan, K. Steffens, R. Zangmeister, J. A. Schreifels and M. V. Rao, Appl. Surf. Sci., 2012, 258, 6056–6063 CrossRef CAS PubMed.
- (a) Y. Kong, Y. Zhong, X. D. Shen, S. Cui, J. J. Zhang and M. Yang, J. Porous Mater., 2013, 20, 845–849 CrossRef CAS; (b) Y. Kong, Y. Zhong, X. D. Shen, S. Cui, M. Yang, K. M. Teng and J. J. Zhang, J. Non-Cryst. Solids, 2012, 358, 3150–3155 CrossRef CAS PubMed; (c) Y. Kong, Y. Zhong, X. D. Shen, S. Cui and M. Fan, Microporous Mesoporous Mater., 2014, 197, 77–82 CrossRef CAS PubMed.
- K. Kaneko, J. Membr. Sci., 1994, 96, 59–89 CrossRef CAS.
- (a) D. J. Boday, P. Y. Keng, B. Muriithi, J. Pyun and D. A. Loy, J. Mater. Chem., 2010, 20, 6863–6865 RSC; (b) K. Kanamori, M. Aizawa, K. Nakanishi and T. Hanada, Adv. Mater., 2007, 19, 1589–1593 CrossRef CAS.
- L. He, M. Fan, B. Dutcher, S. Cui, X. D. Shen, Y. Kong, A. G. Russell and P. McCurdy, Chem. Eng. J., 2012, 189, 13–23 CrossRef PubMed.
- G. Qi, Y. Wang, L. Estevez, X. Duan, N. Anako, A.-H. A. Park, W. Li, C. W. Jones and E. P. Giannelis, Energy Environ. Sci., 2011, 4, 444–452 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11261j |
| This journal is © The Royal Society of Chemistry 2014 |