
Self-assembly amphipathic peptides induce active enzyme aggregation that dramatically increases the operational stability of nitrilase†
Xiaofeng Yanga,
An Huanga,
Jizong Penga,
Jufang Wanga,
Xiaoning Wangc,
Zhanglin Lin*b and
Shuang Li*a
aGuangdong Key Laboratory of Fermentation and Enzyme Engineering, School of Bioscience and Bioengineering, South China University of Technology, Guangzhou Higher Education Mega Center, Panyu District, Guangzhou 510006, China. E-mail: shuangli@scut.edu.cn; Tel: +86 20 3938 0629
bDepartment of Chemical Engineering, Tsinghua University, One Tsinghua Garden Road, Beijing 100084, China. E-mail: zhanglinlin@mail.tsinghua.edu.cn; Tel: +86 10 6277 0304
cState Key Laboratory of Kidney, The Institute of Life Sciences, Chinese PLA General Hospital, Beijing 100853, China
First published on 27th October 2014
Abstract
Oligomeric nitrilase was fused with an amphipathic self-assembly peptide 18A at the C-terminus and expressed in Escherichia coli. The fusion enzyme spontaneously assembled into active aggregates with >90% native nitrilase activity. Much higher specific activities than the native nitrilase (Nit) were recorded for the fusion nitrilase (Nit-SEA) at higher temperatures. The enzyme aggregates were purified through cell lysis and centrifugation led to the facile preparation of immobilized particles (Nit-iSEA). Approximately 86% of the initial nitrilase activity was incorporated into the Ca-alginate entrapment beads. The thermostability of the four kinds of different nitrilase variants showed that the Nit-SEA and Nit-iSEA at 45 °C were about 6.7- and 10-fold more stable than the native nitrilase. The nitrile tolerance was also dramatically improved, no apparent substrate inhibition was observed for Nit-iSEA over the range of 30 to 120 mM mandelonitrile. Additionally, the Nit-iSEA could be recycled 20 times with ∼5% loss in activity.
1. Introduction
Nitriles are highly toxic, mutagenic and widespread in nature. Nitrilase (E. C. 3.5.5.1) can directly hydrolyze nitriles to the corresponding acid and ammonia.1–3 Nitrilases display high chemical specificity and frequent enantioselectivity, which makes them attractive enzymes for the production of fine chemicals and pharmaceutical intermediates. Over the past decades, the production of carboxylic acids by various nitrilase-catalyzing processes has been described.4–6 The success in industrial production of R-mandelic acid (Mitsubishi Rayon, Tokyo, Japan; BASF, Ludwigshafen, Germany) and nicotinic acid (Lonza, Shanghai, China) using nitrilases provides clear evidence of the significant economic potential for this class of enzymes.7,8 Furthermore, a number of industrial processes have combined nitrilase-catalyzed steps with chemical reactions, such as the chemoenzymatic synthesis of atorvastatin (Lipitor) from epichlorohydrin.9,10 Nitrilases are also used in the treatment of toxic industrial effluent and cyanide remediation.11Nitrilase-mediated biocatalysis has undergone rapid development. However, a number of common weaknesses of biocatalysts must be overcome. Achieving higher activities and operational stability under extreme environmental conditions are research hotspots from the industrial application perspective.2,12 Burk et al. evolved a nitrilase to increase the enantiometic excess by 10% with a volumetric productivity of 619 g L−1 d−1 through site saturation mutagenesis.13 Mueller et al. cloned and expressed a nitrilase in Escherichia coli (E. coli), which was shown to be highly thermostable with a half-life of 6 h at 90 °C.14 Using a surface display system, the optimum temperature for the surface-displayed dimeric nitrilase in the cell envelope of E. coli was determined as 45 °C instead of 35 °C.14
To improve enzyme-mediated process efficiency, a few routine methods can be followed:15,16 (i) process optimization through medium engineering or reaction conditions are modification to suit the available biocatalyst, such as immobilization; (ii) genetic engineering of the existing enzymes by rational protein design or random/saturated mutagenesis; and (iii) identification of novel enzymes from natural sources that expand the number of new biocatalysts. Unfortunately, the genetic engineering and novel enzyme discovery methods rely on high-throughput screening techniques, and relationships between sequence, structure and mechanism, which are often labor-intensive and time-consuming. Thus, immobilization appears to be the approach of considerable interest with numerous advantages. However, commercialization of immobilization methods remains limited, because of the associated high costs and storage problems. Thus, research focusing on overcoming the current limitations related to immobilization techniques should lead to an expansion for all-round applications.
Self-assembly peptides (SAPs) are a specific type of peptide that can spontaneously assemble into nanostructures.17 DeGrado's team constructed seven-residue peptides that self-assemble into fibrils, which were able to catalyze p-nitrophenyl acetate hydrolysis in the presence of zinc ions.18 Recently, we reported a short, amphipathic octadecapeptide (18A) that promotes the in vivo assembly of active protein aggregates or active inclusion bodies.19 An additional set of self-complementary peptides was found to drive the target soluble proteins into active inclusion bodies when expressed in E. coli.20,21 These active protein aggregates induced by terminally attached SAPs have been successfully applied in protein purification with high yields and purity.22
Fusion of some proteins or peptides to the N or C terminus of a target enzyme has been reported to achieve improved enzyme performance.23–25 However, this research direction has focused on monomeric proteins, whereas the majority of nitrilases must form oligomers with 4–22 subunits or active spirals of variable lengths to gain catalytic activity.12 In this study, the amphipathic 18A short peptide was introduced to the C terminus of a nitrilase, thereby inducing the active formation of inclusion bodies. The structure of the purified native nitrilase and nitrilase aggregates collected from E. coli cells were observed through transmission electron microscopy (TEM) to understand the characteristic improvements in operational stability when the short peptide was attached. To achieve better thermal stability, toxic substrate tolerance and convenient handling of enzyme catalysis, the nitrilase aggregates were then encapsulated by sodium alginate to immobilize the enzyme aggregates (Fig. 1).
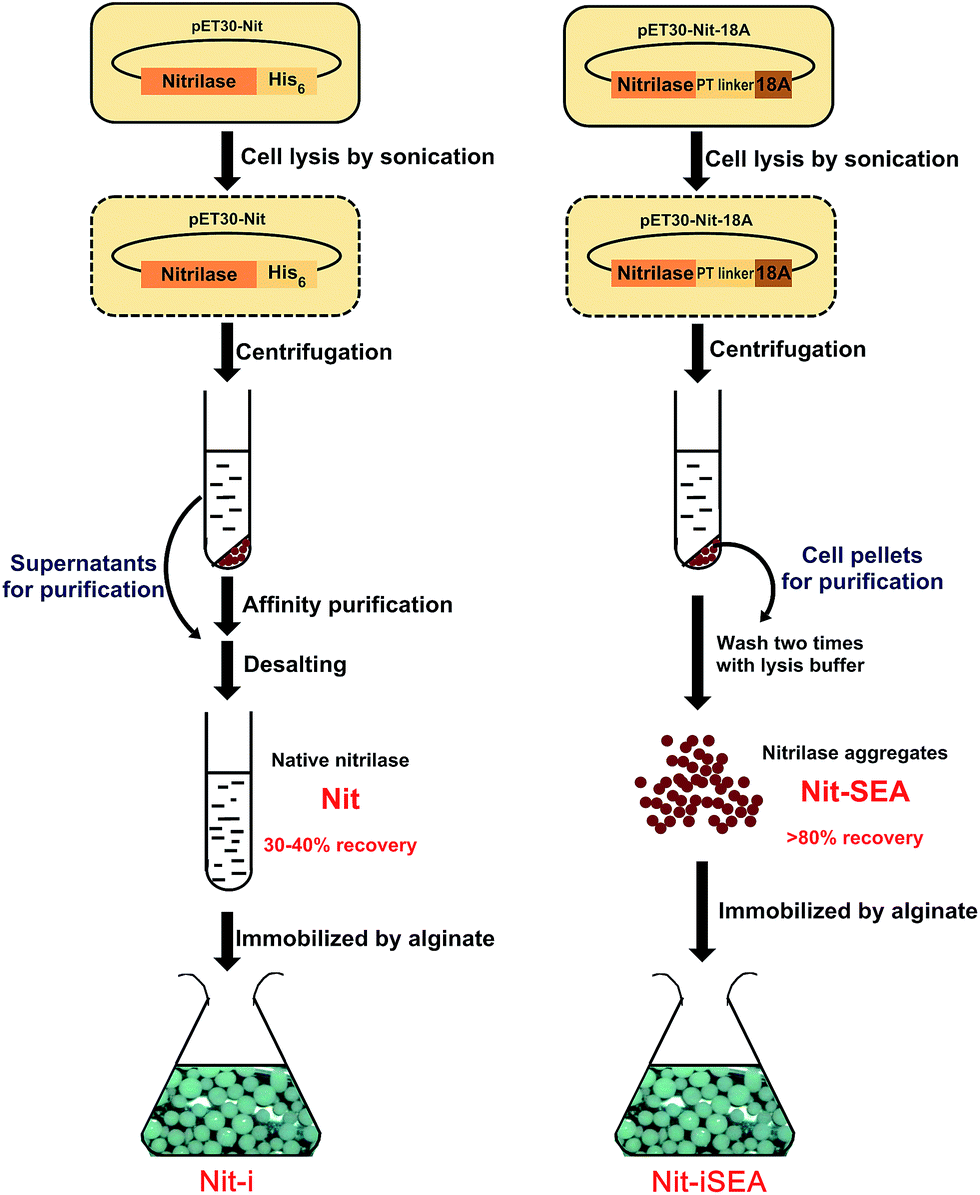 | ||
| Fig. 1 Preparation of different forms of nitrilase, native nitrilase (Nit), 18A fused nitrilase aggregates (Nit-SEA), Ca-alginate immobilized native nitrilase (Nit-i) and Ca-alginate immobilized nitrilase aggregates (Nit-iSEA). | ||
2. Materials and methods
2.1. General remarks
Mandelonitrile and enantiomers of mandelic acid were purchased from Sigma-Aldrich (St. Louis, MI, USA), and nitriles were obtained from TCI (Tokyo, Japan). For genetic manipulation, all enzymes were from Fermentas of Thermo Scientific (Pittsburgh, PA, USA). All solvents were of High Performance Liquid Chromatography (HPLC) grade and obtained from Merck (Darmstadt, Germany). All other chemical reagents used were analytical grade and procured from standard companies.2.2. Construction of expression vectors
Nitrilase gene from Alcaligenes faecalis JM3 (Genbank no. D13419.1) was synthesized by Sangon (Shanghai, China) and amplified using PrimeSTAR® (TaKaRa, Otsu, Japan) with the forward primer 5′-TTAGCCCCATATGCAGACAAGAAAAATCGTCCG-3′ and the reverse primer 5′-TCTGTAAGCTTGGACGGTTCTTGCACCAGTAG-3' (Nde I and Hind III sites are underlined). Purified PCR products were then restriction digested and inserted between the Nde I and Hind III sites of pET30a and pET-c18A19 plasmids to yield the expression vectors pET-Nit and pET-Nit-18A. These two vectors were chemically transformed into E. coli BL21(DE3) competent cells for the expression of native nitrilase (pET-Nit) and the 18A peptide fused nitrilase construct (pET-Nit-18A), respectively. 6× His tag was fused to the native nitrilase in pET-Nit for purification purposes.2.3. Expression of nitrilase
E. coli BL21(DE3) cells with pET-Nit or pET-Nit-18A were grown at 37 °C to the early exponential phase, followed by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM. The cultures were incubated at 22 °C for a further 20 h to express the nitrilase. The harvested cells were collected and resuspended in PB buffer (50 mM sodium phosphate, 50 mM NaCl, pH 7.5), followed by sonication (Scientz JY92-IIN, Ningbo, China) on ice to lyse the cells. The cell lysates were centrifuged (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, 30 min, 4 °C) and the soluble fractions were collected. The precipitates were washed once with PB buffer and resuspended in the same volume of PB buffer. The amount of target protein in both fractions was estimated densitometrically by denaturing polyacrylamide gel electrophoresis (SDS-PAGE) with bovine serum albumin (BSA) as the standard.26 The distributions of enzymatic activities in the soluble and insoluble fractions of the cell lysates were determined as described in the nitrilase activity assay section.
000 × g, 30 min, 4 °C) and the soluble fractions were collected. The precipitates were washed once with PB buffer and resuspended in the same volume of PB buffer. The amount of target protein in both fractions was estimated densitometrically by denaturing polyacrylamide gel electrophoresis (SDS-PAGE) with bovine serum albumin (BSA) as the standard.26 The distributions of enzymatic activities in the soluble and insoluble fractions of the cell lysates were determined as described in the nitrilase activity assay section.
2.4. Purification of nitrilase
For the native nitrilase (Nit) purification, cell pellets were resuspended in 20 mL buffer A (20 mM sodium phosphate, 0.5 M NaCl, 50 mM imidazole, pH 7.4) per gram wet cells and lysed by ultrasonic disruption on ice. After centrifugation (15000 × g, 30 min, 4 °C), the supernatant was passed through a 0.22 μm filter and applied to a HiTrap™ Chelating HP column (GE Healthcare, Piscataway, NJ, USA) and purification followed standard nickel affinity chromatography procedures. The final concentration of native nitrilase was diluted to 5 mg mL−1.
To purify the fusion nitrilase (Nit-SEA), cell pellets were collected and lysed as described before. After centrifugation, the aggregates was washed once with the same volume of Wash Buffer (50 mM sodium phosphate, 50 mM NaCl, 0.8% (v/v) Triton X-100, pH 7.5), and incubated on ice for 20 min. The aggregate was recovered by centrifugation and washed with the same volume of PB buffer and finally diluted in Tris–Cl (50 mM, pH 7.5) to a final concentration of 15 mg mL−1.
For transmission electron microscopy (TEM) observation, the Nit-SEA particles were further purified by differential centrifugation with a sucrose density gradient. The crude Nit-SEA aggregates were resuspended in Ultra-buffer (50 mM NaCl, 20 mM phosphate buffer, 1 mM EDTA, pH 7.5) overlaid on a 12.5 mL gradient of 50–75% (w/v) sucrose prepared in a polyallomer tube (Beckman Coulter, Brea, CA, USA). Gradients were centrifuged at 140000 × g for 2 h at 4 °C using an SW41Ti swinging bucket rotor (Beckman Coulter) on an Optima™ L-100 XP Ultracentrifuge (Beckman Coulter). The buffer of the collected aggregates was replaced with the Ultra-buffer using an Amicon® Ultra-4 centrifugal filter (10 kDa molecular weight cut-off, Millipore, MA, USA).
The purity of the Nit and Nit-SEA was determined by 12% SDS-PAGE.
2.5. Preparation of immobilized enzymes
Two milliliters of the Nit or Nit-18A solution were mixed thoroughly with a 3% sodium alginate solution (Acros Organics, NJ, USA) in the same volume. The enzyme–alginate mixture was dropwise added into a bath of a CaCl2 (0.5 M) solution by syringe. After stirring for an appropriate time on ice, the solution was decanted and the resulting beads (about 1 mm diameter) were washed once with 20 mL of Tris buffer (50 mM Tris-HCl, 50 mM NaCl, pH 7.5). The beads were lyophilized for storage and stored at −20 °C. Before use, the beads were re-swelled with Tris–Cl buffer (50 mM, pH 7.5).For comparison, the Nit and Nit-SEA were also immobilized in similar ways to CLEA®27 using glutaraldehyde (4%, w/v), poyethyleneimine (5%, w/v) or dextran polyaldehyde (5%, w/v) as cross-linking agents.
2.6. Nitrilase activity assay
The colorimetric nitrilase assay was performed in a 96-well plate.28 Five microliters of mandelonitrile (0.5 M) and 25 μL of crude cell lysate were added into 200 μL of the sodium phosphate buffer (10 mM, pH 7.5) containing bromothymol blue (0.01%) (w/v), and incubated in the dark at 45 °C for 3 h. Subsequently, a color change from bluish-green to yellow would be observed if the nitrilase was active.The nitrilase activity was determined through the biotransformation of mandelonitrile in the phosphate buffer for the purified Nit/Nit-SEA or in a Tris–Cl buffer (50 mM, pH 7.5) for the Nit-i/Nit-iSEA, respectively. Fifty micrograms of purified nitrilase or the resulting immobilized forms prepared at the same concentration were added to the reaction buffer and incubated at 37 °C for 20 min. Samples were taken at appropriate times and HCl (1 M) was added to stop the reaction. After centrifugation at 15000 × g for 5 min, the supernatants were analyzed by HPLC. 1 U of the enzyme activity was defined as the amount of enzyme producing 1 μmol of mandelic acid per minute.
2.7. Temperature and pH effects on nitrilase activities
The optimum temperature of Nit and Nit-SEA was examined with mandelonitrile (30 mM) as the substrate in phosphate buffer pH 7.5 under controlled temperatures ranging from 10 to 65 °C during incubation for 20 min.The optimum pH was determined using sodium acetate buffer at pH 4.0–6.0, sodium phosphate buffer at pH 6.0–8.0, Tris–Cl buffer at pH 8.0–9.0 and glycine–NaOH buffer at pH 9.0–10.0 in 50 mM concentrations for the mandelonitrile conversions. The reaction mixtures were incubated at 37 °C for 20 min and analyzed by HPLC.
The enzyme solution in the phosphate buffer pH 7.5 was heated at 45 and 50 °C for different times separately. After heat-treatment, mandelonitrile (30 mM) was added to the enzyme solution and incubated at 37 °C for 20 min. The resulting mixture was then analyzed by HPLC.
2.8. Substrate specificity testing
The specific activities of Nit and Nit-SEA toward different nitriles with structural diversity were determined by quantifying the amount of ammonia released during the catalysis.29 3-Cyanopyridine, benzonitrile, 3-indoleacetonitrile, mandelonitrile, cyclopropanecarbonitrile, 3-hydroxyglutaronitrile, butyronitrile, benzoylacetonitrile and acylonitrile were used as substrates. Reaction mixture containing purified Nit or Nit-SEA (0.02 mg) and different substrates (30 mM) in 1 mL phosphate buffer was incubated at 45 °C for 20 min, respectively.2.9. Nitrilase reusability
Reusability of Nit-i, Nit-SEA and Nit-iSEA was assessed in 1.5 mL centrifuge tubes. The reaction was terminated by pipetting the supernatant and the beads were washed once with an equal volume of phosphate buffer to ensure that the substrate was removed before the next experiment. The reactant and product concentrations were determined by HPLC.2.10. HPLC analysis
Nitrile hydrolysis was determined at 210 nm by HPLC (Waters 2695, Milford, MA, USA) equipped with a photodiode array detector and a C18 column (SunFire, 5 μm, 4.6 mm × 250 mm) using methanol (40%, v/v) and phosphoric acid (0.1%, v/v) as the eluent. The flow rate was 1.0 mL min−1.Enantiomers determination of mandelic acid was achieved on a CHIRALCEL OD-H column (Daicel Chemical Industries, Japan) and detected spectrophotometrically at 228 nm with a flow rate of 0.8 mL min−1 using hexane, isopropanol and trifluoroacetic acid (90:10:0.1, v/v) as the mobile phase. Prior to the assay, the samples were adjusted to pH 1.5 using HCl (6 M) and extracted with an equal volume of ethyl acetate. The extracts were then concentrated and dissolved in the eluent.
2.11. Transmission electron microscopic analysis
Morphometric analysis of the aggregates in E. coli cells was performed by TEM. The cells were collected after 20 h protein expression. After fixing with glutaraldehyde (2.5% w/v) and osmium tetraoxide (2% w/v), a graded-ethanol serial dehydration step was performed. Subsequently, the cells were embedded in epoxy resins, sectioned, stained by a uranyl acetate solution and lead citrate. Finally, the samples were observed with a Hitachi H-7650 (Hitachi, Japan) transmission electron microscope at an accelerating voltage of 80 kV.The structure of the purified Nit and Nit-SEA was also recorded through TEM observation. Five microliters of purified Nit or Nit-SEA (0.2 mg mL−1) was pipetted onto glow discharged, carbon-coated copper grids and incubated at room temperature for 60 s and blotted with filter paper. The grid was placed onto a drop of phosphotungstic acid (PTA, 2% v/v, pH 7.0) for 50 s. Excess PTA was removed and the TEM grid was examined through a Tecnai spirit transmission electron microscope (FEI, USA) operating at 120 kV. Images were captured under a 1k × 1k CCD camera (Olympus, Tokyo, Japan).
2.12. Molecular mass determination
Analytical gel chromatography on a calibrated column (16 × 500 mm) packed with Sephacryl S-300 HR was carried out. The gel bed was equilibrated with a phosphate buffer (20 mM, 150 mM NaCl, pH 7.2). The native nitrilase was applied to the column and the following standard proteins were used for the calibration curve: bovine catalase (232 kDa), chicken egg white conalbumin (75 kDa), hen egg ovalbumin (43 kDa). The enzyme sample (∼2 mg) was subjected to the Bio-Rad BioLogic LP system at a flow rate of 0.5 mL min−1 with the equilibration buffer. The absorbance of the effluent was recorded at 280 nm. The relative molecular mass of nitrilase was calculated by comparison with the relative mobilities of the standard proteins.3. Results and discussion
3.1. Formation of active nitrilase inclusion bodies in E. coli cells
In our previous work, we found that fusion of particular amphipathic short peptides to the terminus of model monomeric proteins induced the formation of active inclusion bodies.19,21,22 Here, the amphipathic 18A (EWLKAFYEKVLEKLKELF) short peptide was attached to the C-terminus of a nitrilase to investigate the formation of inclusion bodies and the impact this process had on the catalytic properties of the nitrilase.The native nitrilase (fused with a 6× His tag for subsequent purification, designated as Nit, 40.3 kDa) was expressed primarily as a soluble protein (Fig. S1A†). The fusion protein (Self-assembly Enzyme Aggregates of Nitrilase, hereafter Nit-SEA, 43.0 kDa) was found to be predominantly (∼95%) in the insoluble fractions, as judged from SDS-PAGE. Using a quick colorimetric assay method,28 the colors of the supernatant from Nit and the precipitant from Nit-SEA both changed from bluish-green to yellow after 3 h incubation (Fig. S1B†), indicating the Nit-SEA was active in the inclusion bodies.
As reported previously,19 the growth of E. coli containing Nit-SEA was dramatically retarded, as judged from cell density measurements. The final OD600 of the fusion protein was about 1.85 ± 0.22, which is significantly less than 5.60 ± 0.35 recorded for the native protein. TEM showed that the Nit-SEA aggregates resided around the periphery of the cell membrane (Fig. S1C and D†), whereas the native nitrilase and conventional inclusion bodies did not present this phenomenon.20 Results were consistent with our previous findings that the positively charged Lys in the 18A sequence resulted in affinity of the peptide to the negatively-charged cell membrane.19
After induced expression, the recombinant E. coli cells were lysed by sonication and the nitrilase was purified (Fig. S2A†). Judging from the SDS-PAGE, the purity of Nit-SEA reached more than 98% after two wash steps with Wash Buffer and PB buffer (see Experimental section), which was comparable to the purity of the native nitrilase purified by affinity chromatography. The results showed that the enzyme recovery rate of the Nit-SEA was up to 84 ± 2%, which is much higher than 33 ± 1% for Nit purified after normal nickel affinity chromatography and desalting purification. Calculated from the enzyme activities in the soluble and insoluble fractions using 30 mM of mandelonitrile as substrate (Fig. S2B†), the aggregated fusion enzyme (Nit-SEA) accounted for 92.6% of the total E. coli-derived nitrilase activity. Using the total activities of the native nitrilase as the respective benchmark, about 83.2% of nitrilase activity was obtained in the form of active inclusion bodies. These results demonstrated that the fusion of the amphipathic octadecapeptide to the C-terminus of the nitrilase has negligible effect on enzyme activity.
3.2. Catalytic properties of Nit-SEA
Using mandelonitrile as the substrate, the overall apparent kinetic parameters of purified Nit and Nit-SEA were determined by directly assaying the R-mandelic acid formation using HPLC, and the results are summarized in Table 1. The purified nitrilase specific activity was 34.1 and 26.4 U mg−1 for Nit and Nit-SEA, respectively. Nit presented a kcat of 60.4 s−1, a Km of 25.6 mM and an overall catalytic efficiency (kcat/Km) of 141.5 min−1 M−1. Relative to the Nit, the aggregate particles of Nit-SEA exhibited a higher Km value and a lower kcat/Km value. It was deduced that the aggregation of nitrilase increased the substrate diffusion resistance and this ultimately led to a decrease in affinity and catalytic efficiency of the biocatalyst towards its substrate. However, the optical purities of R-mandelic acid catalyzed by Nit and Nit-SEA were similar. Although there was no apparent change in the optimum temperature and pH for Nit and Nit-SEA (45 °C, pH 7.5) according to the temperature and pH profiles (Fig. 2), the catalytic activity for Nit-SEA presented higher activity in the higher temperature zone when compared with Nit. For example, the activity of Nit-SEA was 70% at 60 °C relative to the activity at 45 °C, whereas it was only 23% for Nit under similar conditions.| Km [mmol L−1] | Kcat [s−1] | Kcat/Km [mmol L−1 min−1] | Specific activity [U mg−1] | eea [%] | Optimum temperature [°C] | Optimum pH | |
|---|---|---|---|---|---|---|---|
| a Abbreviation of enantiomeric excess of mixtures of R-mandelic acid.b Each value is the average of triplicate experiments and the variation about the mean is below 5%. | |||||||
| Nit | 25.6 | 60.4 | 141.5 | 34.1 | 99.5 | 45 | 7.5 |
| Nit-SEA | 33.0 | 59.3 | 107.9 | 26.4 | 99.6 | 45 | 7.5 |
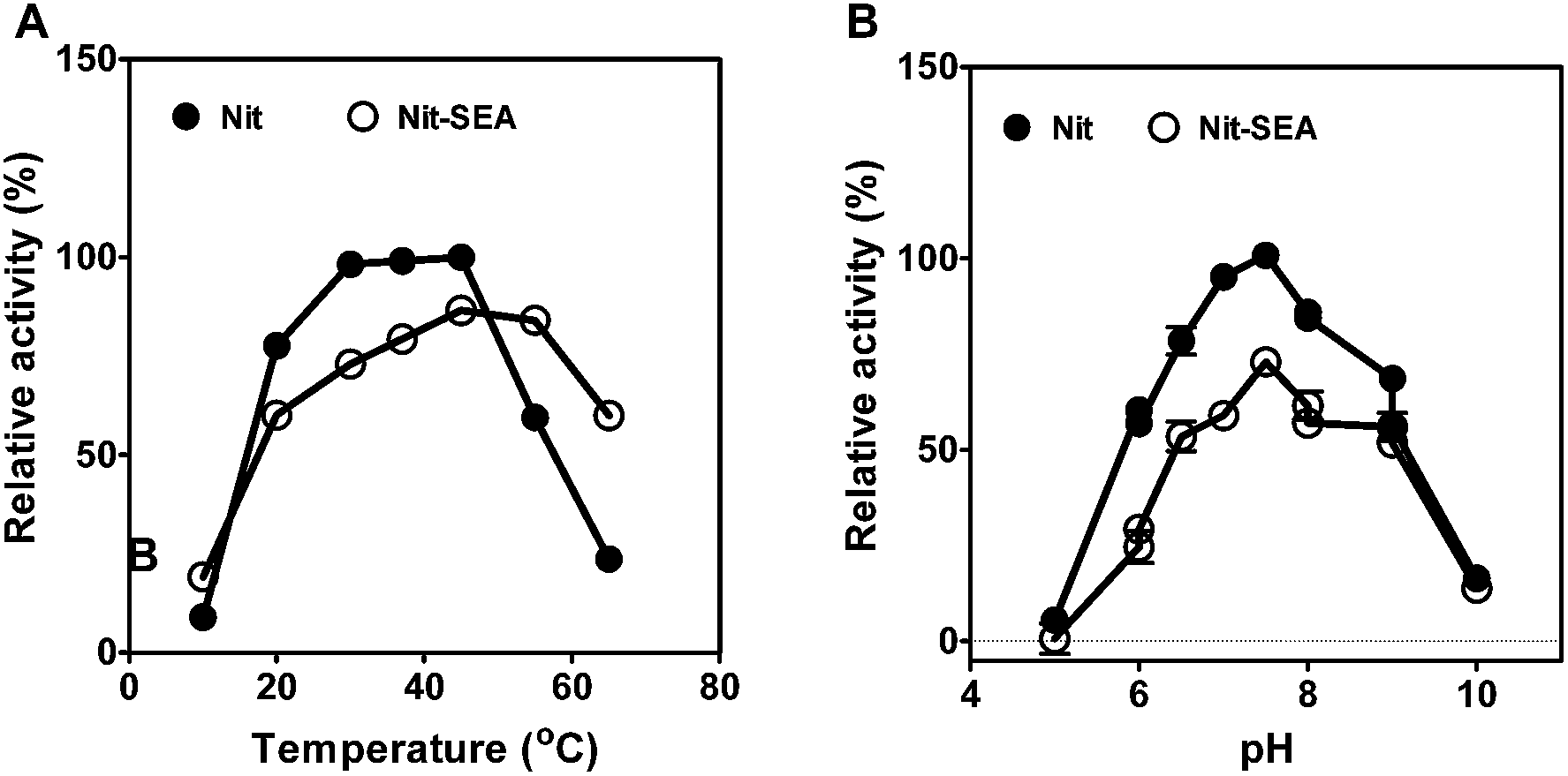 | ||
| Fig. 2 Effects of temperature (A) and pH (B) on the relative nitrilase activities. The specific activity of the native nitrilase at 37 °C and pH 7.5 were taken as 100%. The reactions were performed for 20 min at various temperatures and in the presence of 30 mM mandelonitrile as substrate. The reaction buffers are sodium acetate buffer (pH 4–6), sodium phosphate buffer (pH 6–8), Tris–Cl buffer (pH 8–9) and glycine–NaOH buffer (pH 9–10). | ||
The substrate specificity of the native nitrilase and fusion Nit-SEA were also studied (Fig. 3). Nitrilase activity was determined by the quantification of ammonia released during the reaction.30 Results indicated that the aliphatic nitriles were the preferred substrates for the nitrilase from Alcaligenes faecalis JM3, whereas 3-hydroxyglutaronitrile could not be hydrolyzed. Nitriles directly connected with an aromatic or heterocyclic ring were hydrolyzed at much lower activities than the aliphatic nitriles, except for butyronitrile. Thus, steric hindrance by the side chain of the branched-chain nitriles appears to influence the rate of hydrolysis by the nitrilases. As may be expected, aggregation induced by the amphipathic peptide resulted in the activity decreasing to 50–90% of the native nitrilase activity. For acrylonitrile, the enzyme activities for Nit and Nit-SEA were roughly equal. Surprisingly, the Nit-SEA showed significant activity improvement towards benzoylacetonitrile when compared with the activity of the native Nit.
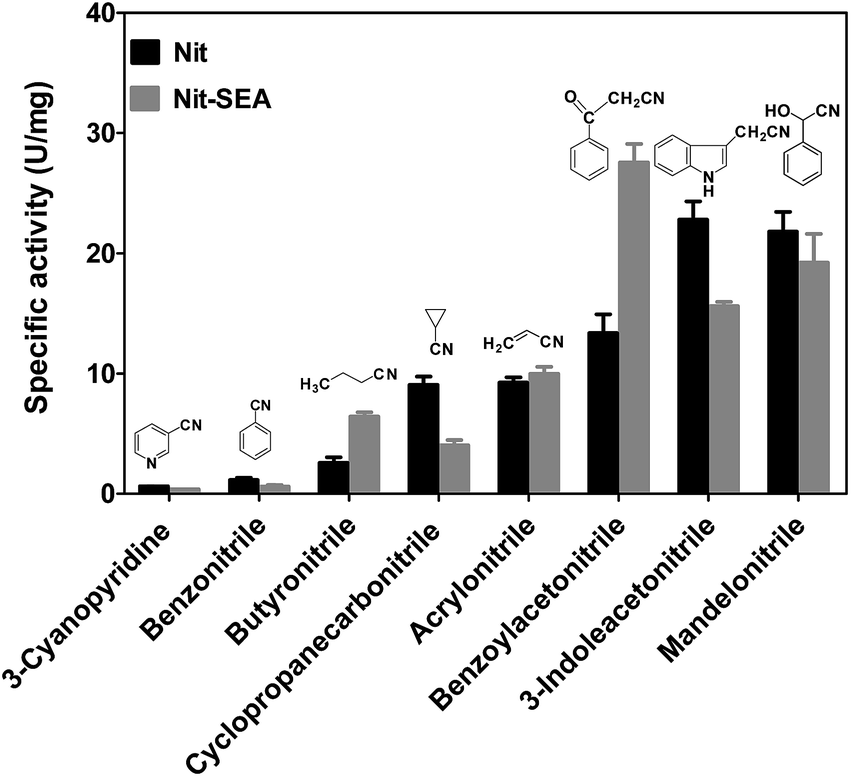 | ||
| Fig. 3 Nitrilase activities toward diverse nitrile compounds with the purified native Nit and fusion Nit-SEA. Data originate from at least three measurements and are presented as the mean ± SD. The reaction mixture, containing 30 mM of each different substrate, 0.02 mg purified nitrilase and 0.95 mL of 50 mM phosphate buffer in a final volume of 1 mL, was incubated at 45 °C for 20 min, and the reaction was terminated by the addition of 0.1 mL of 1 M HCl. The formation of ammonia was determined by HPLC, as described in the Experimental section. The chemical structures are presented above the corresponding chemical names. | ||
3.3. Preparation of immobilized Nit and Nit-SEA
Although there were some improvements in the catalytic characteristics for the short peptide fused nitrilase, we tried to further increase its operational stabilities. Immobilization of nitrilase has been proven to provide some advantages when compared with the non-immobilized enzyme in batch or continuous reactions.31,32 In this study, we first immobilized the purified Nit and Nit-SEA using different concentrations of glutaraldehyde, poly-ethyleneimine and dextran polyaldehyde to cross-link the enzyme. However, recoveries of the enzyme activity were not satisfactory (Table S1†). The highest activity recovered for the immobilized Nit-SEA was obtained as 29.4% cross-linked by 5% dextran polyaldehyde for 4 h.In previous evaluations, alginate encapsulation was demonstrated as a simple, economic and robust method for nitrilase immobilization.33 Thus, we attempted to immobilize the purified Nit and Nit-SEA aggregates with Ca-alginate to further improve the catalytic properties, and these samples were designated as Nit-i and Nit-iSEA. The particle size of Nit-i and Nit-iSEA beads were controlled at about 1 mm in diameter (Fig. S3†). To be hardened, the sodium alginate solution was mixed with particular amines and further cross-linked with glutaraldehyde (Nit-iSEA-G) or dextran polyaldehyde (Nit-iSEA-D) (Fig. 4). However, the glutaraldehyde cross-linking led to a significant loss of nitrilase activity. The highest residual enzymatic activity for Nit-iSEA-G was only 32.0%, obtained following 90 min of immobilization. Using dextran polyaldehyde cross-linking gave similar profiles and the enzyme recovery was nearly identical. Recoveries of the nitrilase activity reached 86.0% and 81.9% for Nit-iSEA and Nit-iSEA-D, respectively, incubated with CaCl2 at 4 °C for 90 min. Sheldon et al.27 also reported that the commonly used cross-linking agent glutaraldehyde gave significant activity loss in penicillin acylase when compared with the use of dextran polyaldehyde. To simplify the operating steps, the nitrilase immobilization was achieved through Ca-alginate entrapment without crosslinking.
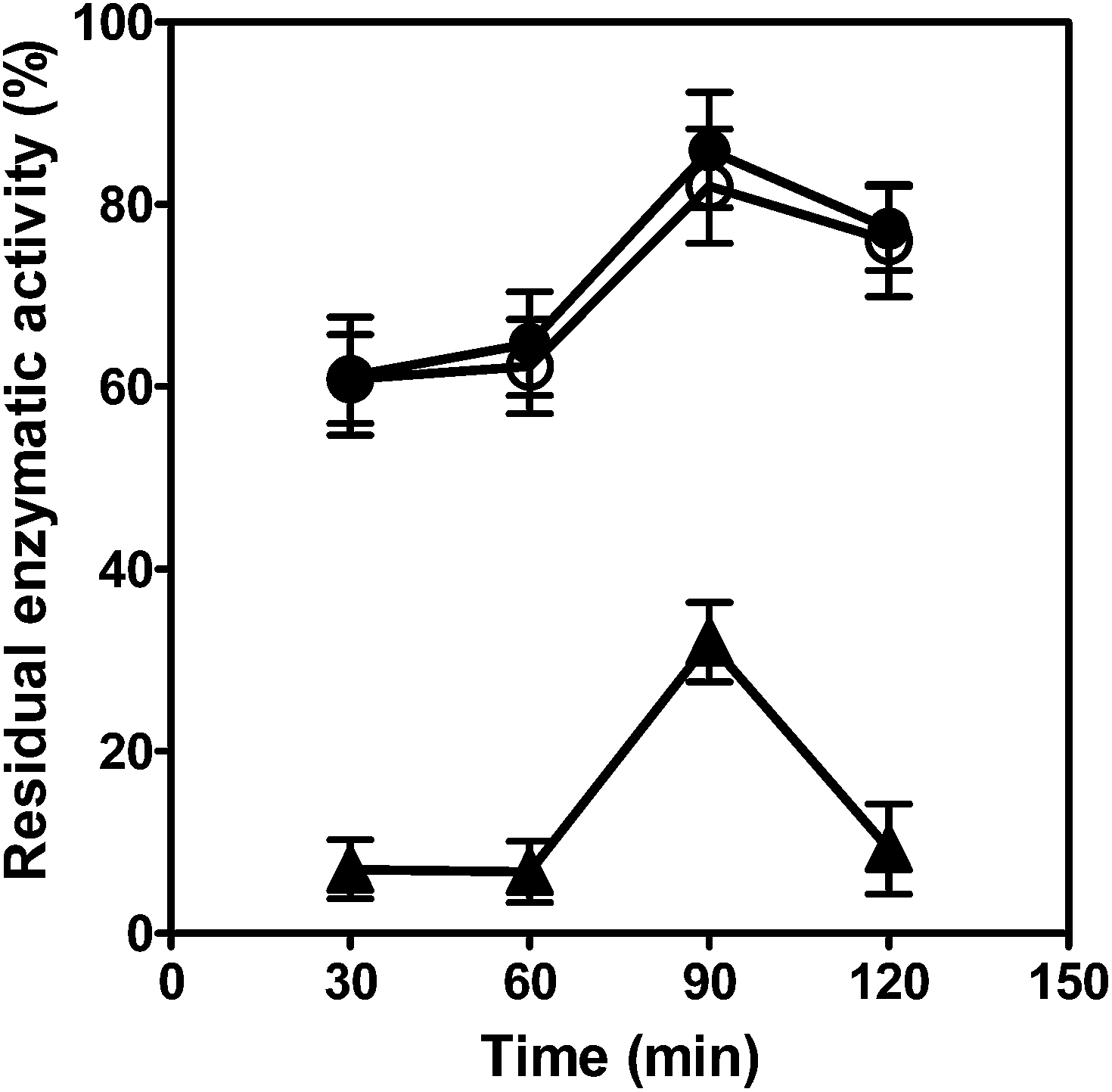 | ||
| Fig. 4 The effect of the crossing linking agent and time on activity recovery for immobilized Nit-SEA. ●, Nit-iSEA, embedded with calcium alginate; ○, Nit-iSEA-D, embedded with calcium alginate and crosslinking through 2.5% (w/v) dextran polyaldehyde; ▲, Nit-iSEA-G, embedded with calcium alginate and crosslinking through 2.5% (w/v) glutaraldehyde. The initial nitrilase activity before immobilization was the benchmark (i.e., 100%). All experiments were performed in triplicate. | ||
3.4. Biocatalyst stability at different temperatures
As shown in the previous results, the relative activity of Nit-SEA at higher temperature was above that of Nit. Here, we investigated the thermal stability of Nit, Nit-i, Nit-SEA and Nit-iSEA (Fig. 5). The results showed that Nit-SEA displayed high thermal stability with half-lives of 161.0 and 56.3 h at 45 and 50 °C, respectively. While for the native nitrilase, the residual activity remained less than 50% following incubation at 45 °C for 23.8 h. At the higher incubation temperature of 50 °C, the half-life of Nit was calculated as 13.1 h. The vast enhancement in the thermal stability may due to the aggregation induced by the 18A attachment, which help to stabilize the conformation of the nitrilase.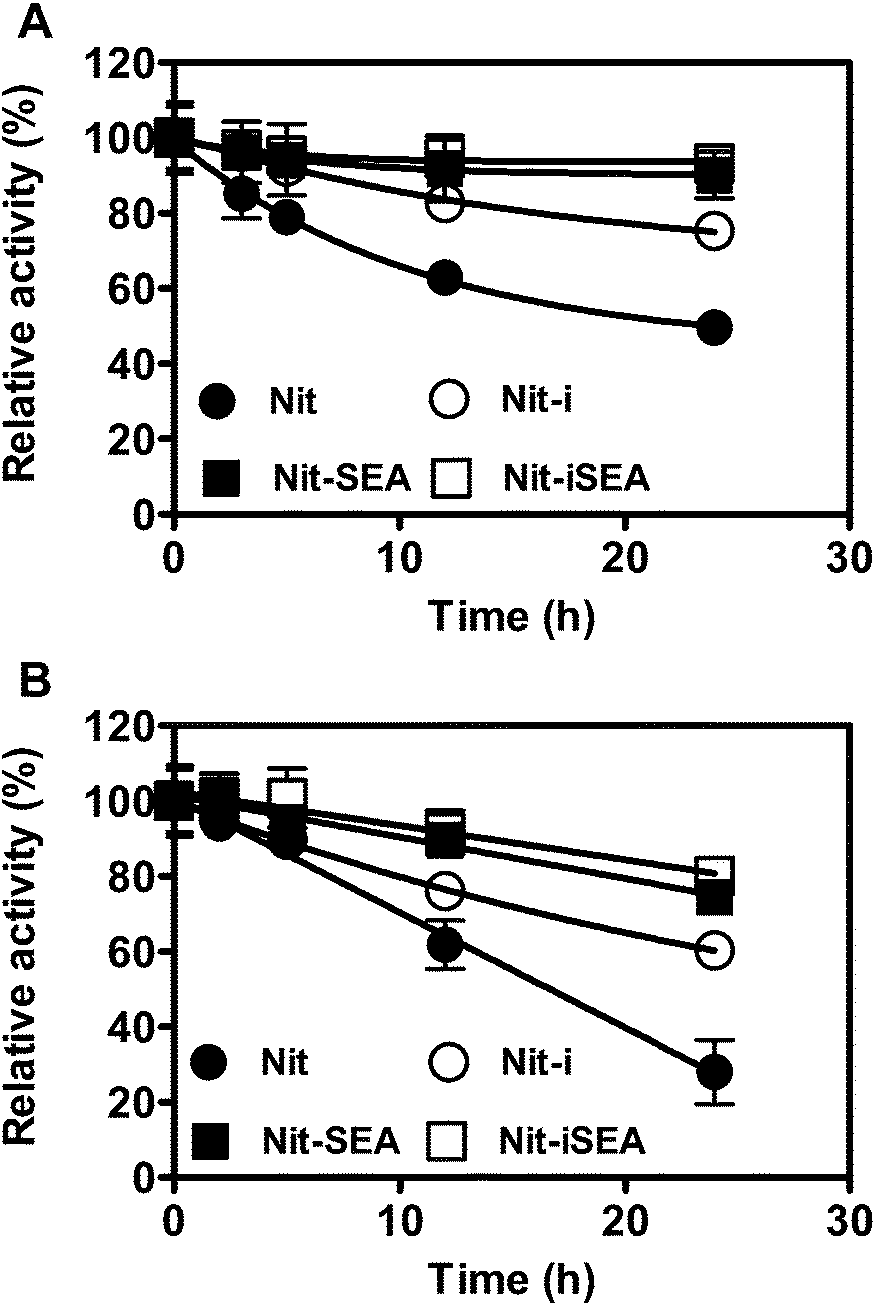 | ||
| Fig. 5 Time profiles of residual nitrilase activities of Nit, Nit-SEA and Nit-iSEA incubated at 45 °C (A) and 50 °C (B) for different periods. ●, Nit; ○, Nit-i; ▲, Nit-SEA; △, Nit-iSEA. The initial enzyme activity of the different forms of the nitrilase was set as 100%. All experiments were carried out in triplicate. | ||
Through Ca-alginate entrapment, the thermal stability of the native and nitrilase aggregates at 45 °C was improved by 2.5- and 1.5-fold relative to the corresponding non-immobilized forms, respectively (Table S2†). It was encouraging that the half-time of the entrapped nitrilase aggregates was dramatically elevated to 237.4 and 75.4 h at 45 and 50 °C, respectively. To emphasize that the nitrilase aggregates were much more stable than the Ca-alginate immobilized native nitrilase under the investigated conditions. All the results demonstrated that enzyme aggregation induced by the amphipathic short peptides followed by alginate embedded immobilization increased nitrilase thermal stability significantly. It has been reported that the self-assembly peptide could enhance the thermal stability of the target enzyme.
Enzyme stabilization is important for any industrial application of enzymes. Immobilization of enzymes is a useful tool to achieve higher thermal stability.34–36 An effective method for enzyme immobilization as CLEA® (cross-linked enzyme aggregates) was developed recently.27,37 P. putida nitrilase has been reported to remain stable for 75 h at 4 °C using the CLEA® technique.32 The CLEA® method involves precipitation of the enzyme from aqueous buffer followed by cross-linking of the enzyme aggregates. In our system, the active protein aggregation is achieved spontaneously within cells without the addition of any agents. The fewer operation steps compared with the CLEA® result in the recovery of higher enzyme activities. Moreover, the successful case of nitrilase prepared by CLEA® reported a maximum activity recovery of 70%, and more than 50% activity was lost after 140 h incubation at 45 °C.32 Compared with our research results, the activity recovery and half-life of the nitrilase-CLEA are not competitive, As stated aformentioned, the activity recovery of Nit-iSEA was about 86% and the half-life of Nit-iSEA was >230 h at 45 °C.
SAPs were also used to improve the catalytic properties of biocatalysts through fusion to the termini of enzymes. By fusing with xylan-binding module CtCBM22A at the N-terminus of a gluco-oligosaccharide oxidase, the catalytic activity of the enzyme was increased 2–3-fold while the thermostability and substrate inhibition were not altered.24 Recently, Zhou et al. reported that the half-life of nitrile hydratase was successfully increased ∼1.5-fold at 50 °C through fusion with the ELK16 peptide.25 Additionally, the thermal stability of a lipoxygenase was also improved from 10 to 46 min at 50 °C via fusion of a SAP to the N terminus.38 All these reported results focused on monomeric proteins and involved the fusion of a SAP with an enzyme without further follow-up research.
3.5. Effect of mandelonitrile concentration on nitrilase activity
Nitrile compounds are highly toxic and harmful to nitrilases, which makes the substrate concentration a limiting factor in the biotransformation process. However, a higher substrate concentration tolerance could give rise to a higher reaction rate.39 In the presence of different mandelonitrile concentrations, a typical substrate inhibition effect was observed for the native nitrilase from Alcaligenes faecalis JM3 (Fig. 6). The highest nitrilase activity was recorded at 30 mM mandelonitrile for Nit. Nevertheless, this was increased to 50 mM for Nit-SEA and Nit-i and 60 mM for Nit-iSEA. The substrate inhibition of the native nitrilase resulted in lowering of the mandelonitrile transformation rate to 26.9% at 60 mM. After immobilization of the native nitrilase or induced aggregation by 18A, the residual nitrilase activity was 45 and 63% at 70 mM mandelonitrile, respectively. Importantly, the immobilized particles of Nit-iSEA still retained over 80% nitrilase activity when the mandelonitrile concentration was increased to 120 mM. No apparent substrate inhibition was observed for Nit-iSEA over the mandelonitrile concentration range of 10–120 mM. According to Copeland,40 substrate inhibition can be accounted for by eqn (1).
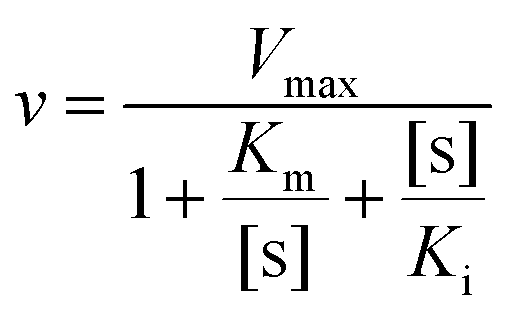 | (1) |
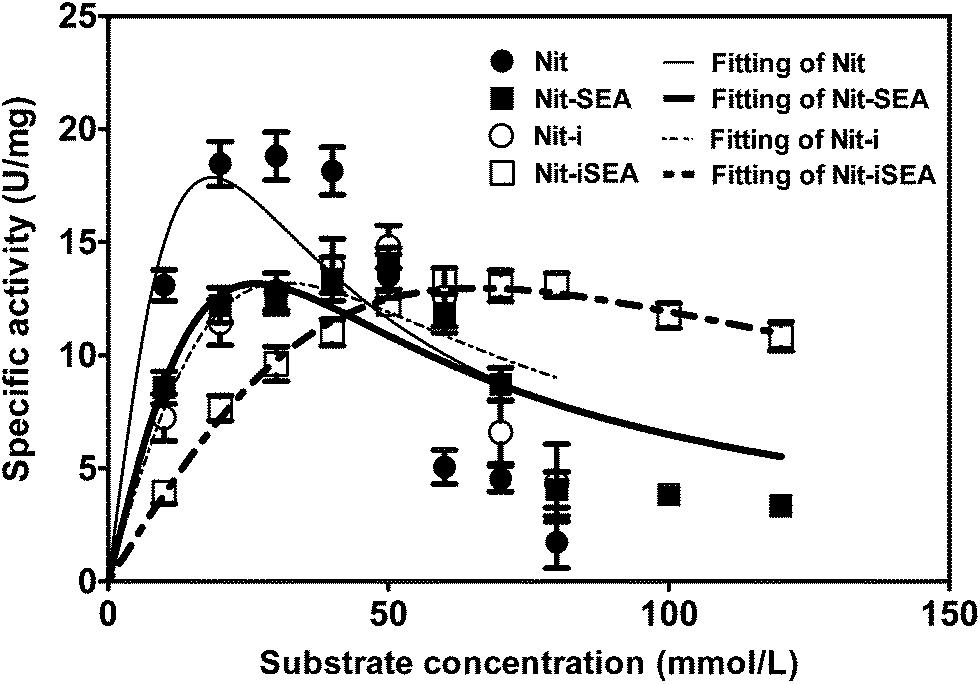 | ||
| Fig. 6 Comparison of substrate tolerance between the native nitrilase, nitrilase-18A, immobilized nitrilase and immobilized nitrilase-18A with 0.5 M calcium alginate. The specific activities of the different forms of nitrilase assayed using different concentrations of mandelonitrile as the substrate. The values reported correspond to the mean of experiments repeated in triplicate. | ||
3.6. Repeated use capability
The Nit-i, Nit-SEA and Nit-iSEA were used to convert 30 mM mandelonitrile to R-mandelic acid. After 100% conversion, the beads/aggregates were recovered by centrifugation for a subsequent round of bioconversion. The native nitrilase was not run in recycle reactions, because it cannot be recycled using conventional methods. After eight such recycles, the immobilized native nitrilase (Nit-i) lost more than 70% of the initial nitrilase activity (Fig. 7). However under similar conditions, Nit-SEA retained about 80% of its original activity. Even more encouraging was the observation that the Nit-iSEA beads specific activity remained >95% of the initial specific activity after 20 batches of consecutive conversion. The results showed that the activity of the free and immobilized nitrilase aggregates were stable in the batch reactions. It should be noted that time required to catalyze the 100% conversion of mandelonitrile by Nit-iSEA was only extended from 50 to 60 min for the 20th batch reaction compared to the first reaction. However in the 8th batch reaction, the mandelonitrile was still not completely converted to mandelic acid catalyzed by Nit-i after 150 min incubation. Therefore, the fusion of an amphipathic short peptide to the C-terminus of the nitrilase efficiently improved the catalysis stability for the bioconversion of mandelonitrile to R-mandelic acid. Compared with the free catalyst, separating the aggregated or immobilized biocatalysts from the reaction mixture is easier and readily facilitates the repeated batch operations. The entrapment of cells harboring A. facilis nitrilase was shown previously to be reusable for 10 batches, whereas free cells completely lost activity after nine batches.36 Immobilized Streptomyces nitrilase in an agar–agar matrix can be reused for 25 cycles with an approximately 20% enzyme activity loss.35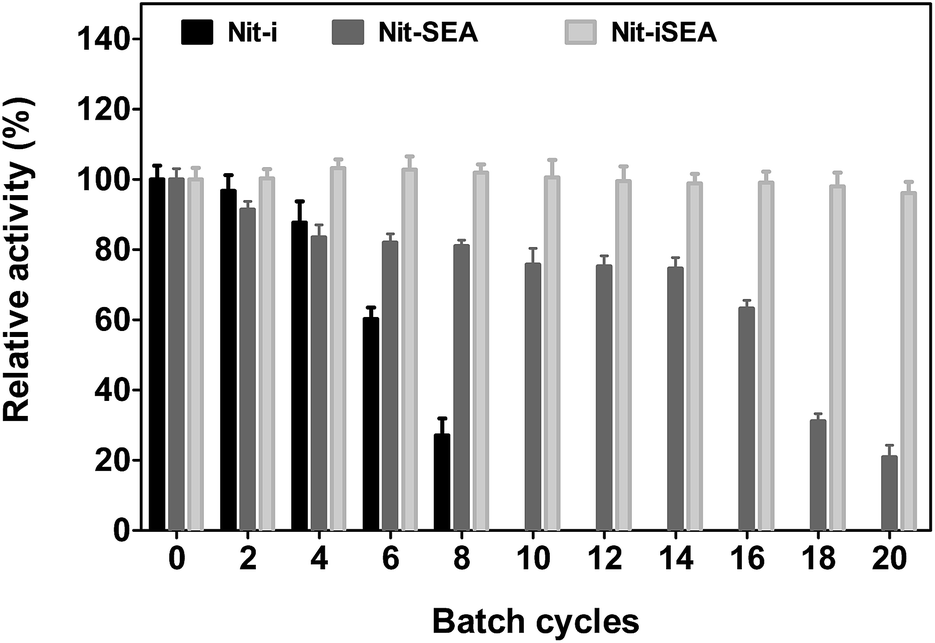 | ||
| Fig. 7 Comparison of the reusability of Nit-i, Nit-SEA and Nit-iSEA. The relative activities are expressed as the percentage of the initial conversion of the reaction by different forms of the nitrilase. | ||
3.7. Structure analysis of the Nit and Nit-SEA
Negative stained transmission electron micrographs showed homo-oligomeric structures for the freshly prepared native nitrilase from Alcaligenes faecalis JM3 (Fig. 8A). A large number of “C” shaped and a few closed ring-like structures were present. The molecular mass of the freshly purified native nitrilase was determined to be about 242 kDa. This suggested that the active forms of the cloned nitrilase were mostly hexamers. A nitrilase from the same strain has also been found to self-associate to form species of 260 kDa.41 The present results are consistent with previous observations that nitrilases readily self associate to form large aggregates of 4–26 subunits.12,42 After storage at 4 °C for 1 month, the enzyme was separated by gel-filtration chromatography into fractions containing an active 242 kDa oligomer and an inactive fraction in the void volume indicating a mass >1 MDa. Using negative-stain electron microscopy, the long-term preservation of Nit was shown to be predominantly amorphous aggregation structures (Fig. 8B).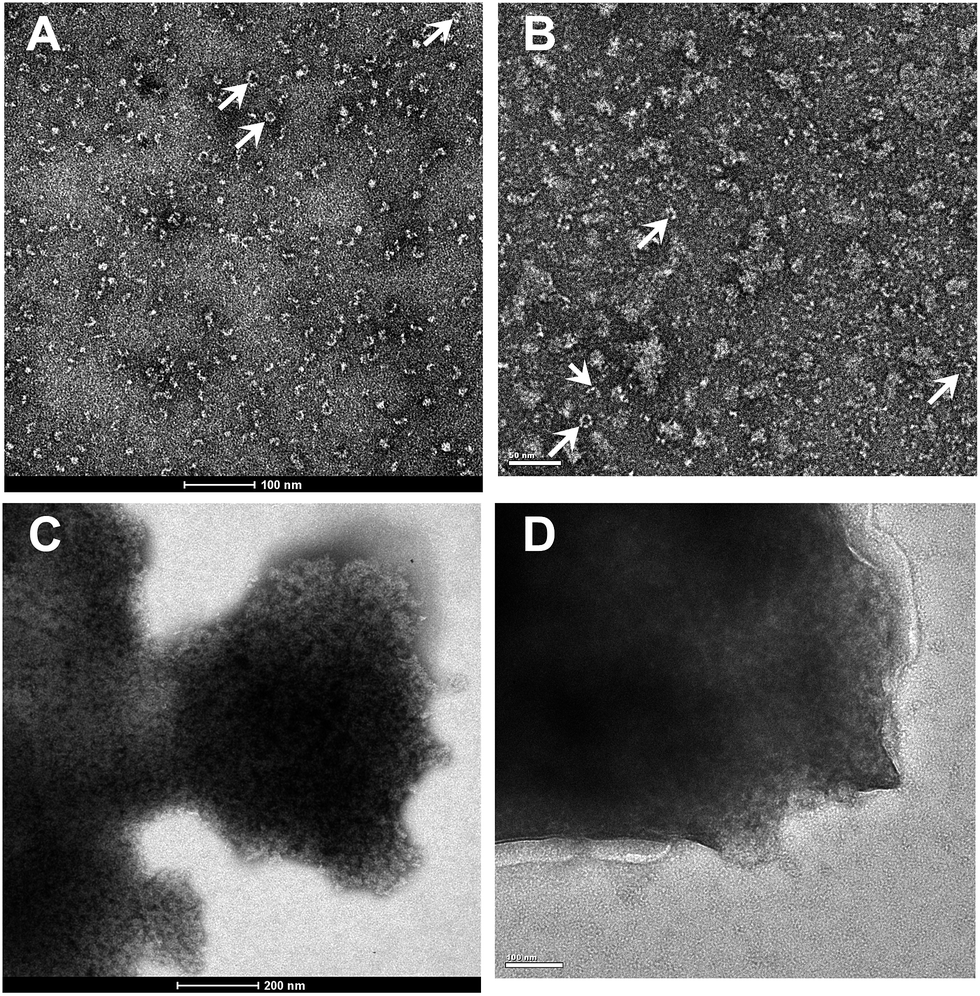 | ||
| Fig. 8 Negative stained transmission electron micrographs (TEM) of the purified native nitrilase and 18A induced nitrilase aggregates. (A) Freshly purified native nitrilase. The “C” shaped and closed ring-like (indicated by arrows) structures with a diameter of ∼10 nm are presented. (B) Purified native nitrilase incubated at 4 °C for 1 month. Amorphous aggregation structures are the major form. The nitrilase structures are indicated by arrows. (C) Freshly purified nitrilase aggregates isolated by sucrose density gradient centrifugation. They are electron-dense aggregates. (D) Purified nitrilase aggregates were incubated at 4 °C for 1 month. The samples were negatively stained with 2.0% (v/v) phosphotungstic acid and observed under a FEI Tecnai 20 transmission microscope operating at 120 kV. Scale bars are indicated at the bottom of each image. | ||
Some of apolipoprotein A-I or other small α-helical apolipo-proteins are associated with forms of amyloidosis in human.43 18A, an apolipoprotein A–I, has a strong helical tendency and lipid-associating affinity. 18A can self-assemble by coiled-coil in the aqueous solution, and form discoidal particles44,45 or helix fibrils.46 A previous study reported that the LipA-18A appeared to adopt a mesh-like structure after limited proteolytic treatment by protease K, as observed by TEM operating at 75 kV.19 In this study, Nit-SEA particles were ultra-purified by differential centrifugation with a sucrose density gradient, and presented roughly the same structures with LipA-18A, but totally different with the native nitrilase (Fig. 8C). It should be noted that the Nit-SEA showed negligible change in activity and structure when stored at 4 °C for 1 month (Fig. 8D). The aggregates provide a concentration of active sites that can be purified and immobilized easily, also avoided the amorphous self-aggregation of native nitrilase, which may lead to activity loss when stored for long periods.
4. Conclusions
We demonstrated that the self-assembly peptide 18A could be employed for the formation of oligomeric nitrilase active inclusion bodies in E. coli. A greater reaction rate than the native nitrilase was observed for the active nitrilase aggregates at temperatures of 55 and 65 °C. Dramatic improvements in the substrate tolerance, operational stability and recycle times were successfully achieved through coupling the fusion of 18A to the nitrilase with alginate entrapment. Owing to the simplicity of the aggregation and purification process with high residual enzyme activity and excellent operational stability, the 18A induced self-assembly active aggregates should be potentially suitable in industrial biocatalyst development.Acknowledgements
We would like to thank the Center for Biological Imaging (CBI), Institute of Biophysics, Chinese Academy of Science for technical support in electron microscopy and we are grateful to Dr Xiaojun Huang and Dr Gang Ji for their help in preparing the TEM samples and collecting the TEM images. This work was supported in part by the National Natural Science Foundation of China (21276096), National High-tech R&D Program of China (863 Program, 2012AA022205), Science and Technology Planning Project of Guangdong, China (2011B020309005), Foundation for Distinguished Young Talents in Higher Education of Guangdong, China (LYM08012), the CAS Key Laboratory of Microbial Physiological and Metabolic Engineering, Institute of Microbiology, Chinese Academy of Sciences (KLIM-201301), and the Open Project Program of Guangdong Key Laboratory of Fermentation and Enzyme Engineering, SCUT (FJ2013006). Shuang Li was funded by the Pearl River New-Star of Science & Technology supported by Guangzhou City (2012J2200012).References
- C. Kiziak, D. Conradt, A. Stolz, R. Mattes and J. Klein, Microbiology-SMG, 2005, 151, 3639–3648 CrossRef CAS PubMed.
- J.-S. Gong, Z.-M. Lu, H. Li, J.-S. Shi, Z.-M. Zhou and Z.-H. Xu, Microb. Cell Fact., 2012, 11, 142 CrossRef CAS PubMed.
- H. C. Pace and C. Brenner, Genome Biol., 2001, 2, 1–9 CrossRef.
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- L. Martinkova, V. Vejvoda, O. Kaplan, D. Kubac, A. Malandra, M. Cantarella, K. Bezouska and V. Kren, Biotechnol. Adv., 2009, 27, 661–670 CrossRef CAS PubMed.
- D. Robert, in Biocatalysis in the pharmaceutical and biotechnology industries, ed. R. N. Patel, CRC Press, London, 2006, ch. 1, pp. 1–20 Search PubMed.
- N. M. Shaw, K. T. Robins and A. Kiener, Adv. Synth. Catal., 2003, 345, 425–435 CrossRef CAS.
- D. Brady, A. Beeton, J. Zeevaart, C. Kgaje, F. van Rantwijk and R. A. Sheldon, Appl. Microbiol. Biotechnol., 2004, 64, 76–85 CrossRef CAS PubMed.
- S. Bergeron, D. A. Chaplin, J. H. Edwards, B. S. W. Ellis, C. L. Hill, K. Holt-Tiffin, J. R. Knight, T. Mahoney, A. P. Osborne and G. Ruecroft, Org. Process Res. Dev., 2006, 10, 661–665 CrossRef CAS.
- M. Müller, Angew. Chem., Int. Ed., 2005, 44, 362–365 CrossRef PubMed.
- L. J. Basile, R. C. Willson, B. T. Sewell and M. J. Benedik, Appl. Microbiol. Biotechnol., 2008, 80, 427–435 CrossRef CAS PubMed.
- R. N. Thuku, D. Brady, M. J. Benedik and B. T. Sewell, J. Appl. Microbiol., 2009, 106, 703–727 CrossRef CAS PubMed.
- G. DeSantis, K. Wong, B. Farwell, K. Chatman, Z. L. Zhu, G. Tomlinson, H. J. Huang, X. Q. Tan, L. Bibbs, P. Chen, K. Kretz and M. J. Burk, J. Am. Chem. Soc., 2003, 125, 11476–11477 CrossRef CAS PubMed.
- P. Mueller, K. Egorova, C. E. Vorgias, E. Boutou, H. Trauthwein, S. Verseck and G. Antranikian, Protein Expression Purif., 2006, 47, 672–681 CrossRef CAS PubMed.
- S. Li, X. Yang, S. Yang, M. Zhu and X. Wang, Comput. Struct. Biotechnol. J., 2012, 2, e201209017 Search PubMed.
- K. Steiner and H. Schwab, Comput. Struct. Biotechnol. J., 2012, 2, e201209010 Search PubMed.
- S. Maude, L. R. Tai, R. P. Davies, B. Liu, S. A. Harris, P. J. Kocienski and A. Aggeli, Top. Curr. Chem., 2012, 310, 27–69 CrossRef CAS.
- C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stohr, T. A. Smith, X. Z. Hu, W. F. DeGrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303–309 CrossRef CAS PubMed.
- Z. Lin, B. Zhou, W. Wu, L. Xing and Q. Zhao, Faraday Discuss., 2013, 166, 243–256 RSC.
- W. Wu, L. Xing, B. Zhou and Z. Lin, Microb. Cell Fact., 2011, 10, 9 CrossRef CAS PubMed.
- B. H. Zhou, L. Xing, W. Wu, X. E. Zhang and Z. L. Lin, Microb. Cell Fact., 2012, 11, 10 CrossRef CAS PubMed.
- L. Xing, W. Wu, B. Zhou and Z. Lin, Microb. Cell Fact., 2011, 10, 42 CrossRef CAS PubMed.
- Y. Wang, D. E. Prosen, L. Mei, J. C. Sullivan, M. Finney and P. B. Vander Horn, Nucleic Acids Res., 2004, 32, 1197–1207 CrossRef CAS PubMed.
- V. V. Thu and R. M. Emma, PLoS One, 2014, 9, e95170 Search PubMed.
- Y. Liu, W. Cui, Z. Liu, Y. Cui, Y. Xia, M. Kobayashi and Z. Zhou, J. Biosci. Bioeng., 2014, 118, 249–252 CrossRef CAS PubMed.
- S. Peternel, J. Grdadolnik, V. Gaberc-Porekar and R. Komel, Microb. Cell Fact., 2008, 7, 34 CrossRef PubMed.
- C. Mateo, J. M. Palomo, L. M. van Langen, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2004, 86, 273–276 CrossRef CAS PubMed.
- A. Banerjee, P. Kaul, R. Sharma and U. C. Banerjee, J. Biomol. Screening, 2003, 8, 559–565 CrossRef CAS PubMed.
- M. W. Weatherburn, Anal. Chem., 1967, 39, 971–974 CrossRef CAS.
- W. J. Park, V. Kriechbaumer, A. Moller, M. Piotrowski, R. B. Meeley, A. Gierl and E. Glawischnig, Plant Physiol., 2003, 133, 794–802 CrossRef CAS PubMed.
- A. Panova, L. I. Mersingera, Q. Liu, T. Foo, D. C. Roe, W. L. Spillan, A. E. Sigmund, A. Ben-Bassat, L. W. Wagner, D. P. O'Keefe, S. Wu, K. L. Petrillo, M. S. Payne, S. T. Breske, F. G. Gallagher and R. DiCosimo, Adv. Synth. Catal., 2007, 349, 1462–1474 CrossRef CAS.
- S. Kumar, U. Mohan, A. L. Kamble, S. Pawar and U. C. Banerjee, Bioresour. Technol., 2010, 101, 6856–6858 CrossRef CAS PubMed.
- L. J. Mersinger, E. C. Hann, F. B. Cooling, J. E. Gavagan, A. Ben-Bassat, S. Wu, K. L. Petrillo, M. S. Payne and R. DiCosimo, Adv. Synth. Catal., 2005, 347, 1125–1131 CrossRef CAS.
- P. Kaul, A. Banerjee and U. C. Banerjee, Biomacromolecules, 2006, 7, 1536–1541 CrossRef CAS PubMed.
- V. K. Nigam, A. K. Khandelwal, R. K. Gothwal, M. K. Mohan, B. Choudhury, A. S. Vidyarthi and P. Ghosh, J. Biosci., 2009, 34, 21–26 CrossRef CAS PubMed.
- Z. Q. Liu, M. Zhou, X. H. Zhang, J. M. Xu, Y. P. Xue and Y. G. Zheng, J. Mol. Microbiol. Biotechnol., 2012, 22, 35–47 CrossRef CAS PubMed.
- R. A. Sheldon, Biochem. Soc. Trans., 2007, 35, 1583–1587 CrossRef CAS PubMed.
- X. Y. Lu, S. Liu, D. X. Zhang, X. M. Zhou, M. Wang, Y. Liu, J. Wu, G. C. Du and J. Chen, Appl. Microbiol. Biotechnol., 2013, 97, 9419–9427 CrossRef CAS PubMed.
- D. Costes, G. Rotcenkovs, E. Wehtje and P. Adlercreutz, Biocatal. Biotransform., 2001, 19, 119–130 CrossRef CAS.
- R. A. Copeland, in Enzymes: A practical introduction to structure, mechanism, and data analysis, John Wiley & Sons, Inc., 2004, ch. 5, pp. 109–145 Search PubMed.
- T. Nagasawa, J. Mauger and H. Yamada, Eur. J. Biochem., 1990, 194, 765–772 CrossRef CAS PubMed.
- A. Banerjee, R. Sharma and U. C. Banerjee, Appl. Microbiol. Biotechnol., 2002, 60, 33–44 CrossRef CAS PubMed.
- G. Merlini and V. Bellotti, N. Engl. J. Med., 2003, 349, 583–596 CrossRef CAS PubMed.
- G. M. Anantharamaiah, J. I. Jones, C. G. Brouillette, C. F. Schmidt, B. H. Chung, T. A. Hughes, A. S. Bhown and J. P. Segrest, J. Biol. Chem., 1985, 260, 248–255 Search PubMed.
- R. M. Epand, A. Gawish, M. Iqbal, K. B. Gupta, C. H. Chen, J. P. Segrest and G. M. Anantharamaiah, J. Biol. Chem., 1987, 262, 9389–9396 CAS.
- K. L. Lazar, H. Miller-Auer, G. S. Getz, J. P. R. O. Orgel and S. C. Meredith, Biochemistry, 2005, 44, 12681–12689 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11236a |
| This journal is © The Royal Society of Chemistry 2014 |