
Synthesis and characterization of TiO2 nanotube supported Rh-nanoparticle catalysts for regioselective hydroformylation of vinyl acetate†
Yukun Shi,
Xiaojing Hu,
Baolin Zhu,
Shurong Wang,
Shoumin Zhang and
Weiping Huang*
Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), The Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Tianjin Key Lab of Metal and Molecule-based Material Chemistry and College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: hwp914@nankai.edu.cn; Fax: +86-22-23502669; Tel: +86-22-23502996
First published on 12th November 2014
Abstract
Three procedures: the impregnation-borohydride reduction procedure, the impregnation-alcohol reduction procedure and the impregnation-photoreducing procedure, were utilized for preparing TiO2 nanotube supported rhodium nanoparticle catalysts, in which rhodium acetate was used as a rhodium source. Catalysts were characterized with TEM, ICP, XPS and XRD; their catalytic performances for hydroformylation of vinyl acetate were evaluated. Of these catalysts prepared by three different methods, the catalyst prepared by the impregnation-photoreducing procedure showed the highest catalytic activity under the same reaction conditions. The effects of pressure of syngas, solvents, temperature, Rh content and reaction time on the hydroformylation were examined in detail. Under the optimized reaction conditions, the conversion of vinyl acetate can reach at 100%; the chemoselectivity for aldehydes was 74.70% and the regioselectivity for the branched aldehyde was 95%.
1. Introduction
There are several methods for preparing aldehydes, but the dominant technology is catalytic hydroformylation, which is an elegant method of preparing a wide range of aldehydes in a single step by the reaction of alkenes, CO and H2 with high selectivity.1 The reaction was first discovered by Otto Roelen in 1938 during investigations on the formation of oxygenated products in cobalt-catalyzed Fisher–Tropsch reactions. Several million tons of aldehyde, world wide, are produced annually via the hydroformylation reaction, most of which are reduced to alcohols or oxidized to carboxylic acids. However, regioselective hydroformylation of functionalized alkenes, e.g. vinyl acetate, is still a challenging topic and has drawn great attention.2–6 With the regioselective hydroformylation of functionalized alkenes, target products having functional groups at different positions can be synthesized.7,8 For example, the hydroformylation of vinyl acetate provides bifunctional products (Scheme 1), which can be further converted into synthetically useful compounds, e.g. lactic acid and so on. To date, though there are few reports concerning hydroformylation of functionalized alkenes with high regioselectivity carried out under mild conditions, a lot of effort has been made to increase the regioselectivity with high reaction rate in the past decades.3,9 The key to achieve high regioselectivity for target product is to create a novel catalyst.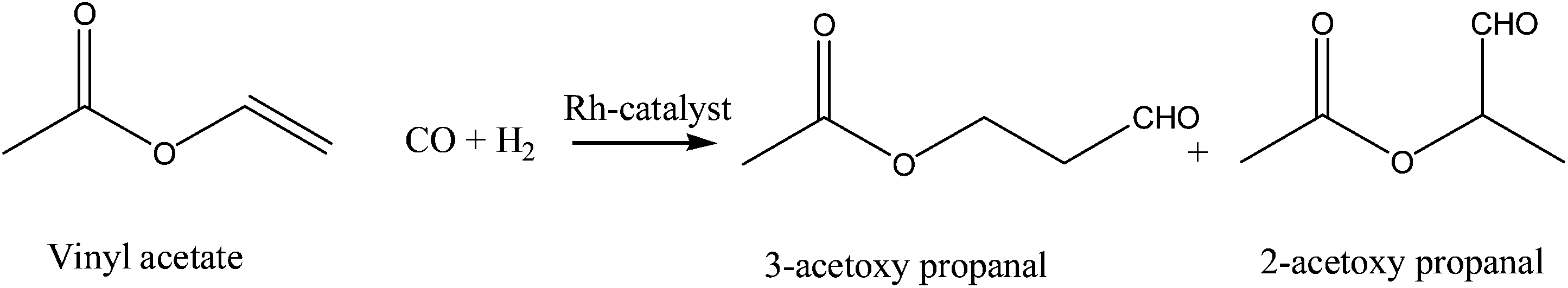 | ||
| Scheme 1 Hydroformylation of vinyl acetate. | ||
Some transition metal-based catalysts involving Rh, Ir, Pd, Co and Ru have been widely used for the hydroformylation reaction.10–13 Due to the lower reaction temperature and pressure, Rh-based catalysts are usually preferred for hydroformylation reaction. Nanoscale Rh particles have been holding people's interests14 for their novel electronic, magnetic, optical and catalytic properties.15–20 Their enhanced performances, especially for size-dependent catalytic properties, result from their size or shape and high surface-to-volume ratio.21 However, pristine Rh nanoparticles are thermally unstable and tend to agglomerate in solution when heated. In order to stabilize the Rh nanoparticles, polymers and surfactants are often used.22–26 However, Sayed's group and Korgel's group have reported that surfactants or capping ligands used in the synthesis of metallic nanoparticles can diminish the catalytic activity of the produced metallic nanocatalysts. Somorjai and co-workers have also reported that poly vinylpyrrolidone (PVP), the most widely used capping agent in size-tunable and shaped nanoparticles synthesis, interacted strongly with Rh nanoparticles resulting in a significant capping agent effect.27–29 Though Rh nanoparticles can be synthesized by using capping agents and subsequently deposited on the high-surface-area solid support, completely removing the capping agents to expose the catalytically “clean” metal surfaces is very difficult. Therefore, developing simple, rapid and surfactant-free approaches for synthesizing Rh nanoparticles deposited on the high surface area solid supports is highly desirable for studying Rh-based catalysts.30,31 Thus, TiO2 nanotube (TNTs), a large surface area inorganic material, has been widely used as support for catalytic materials.32–37
It has been reported that TNTs prepared via hydrothermal treatment are constructed by rolling up one (1 0 1) layer of the anatase structure along the [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 01] direction and the (1 0 1) facet constructed space between walls is about 0.76 nm.38 Such multiwall structure makes TNTs have large specific surface area and nano-confine effect, which can be utilized for stabilizing Rh nanoparticles deposited on the surfaces of TNTs. It is optimistically anticipated that multiwall TNTs supported Rh-nanoparticle catalysts would be very stable, based on our previous work, 37 and have a good catalytic activity for the hydroformylation of alkenes.
01] direction and the (1 0 1) facet constructed space between walls is about 0.76 nm.38 Such multiwall structure makes TNTs have large specific surface area and nano-confine effect, which can be utilized for stabilizing Rh nanoparticles deposited on the surfaces of TNTs. It is optimistically anticipated that multiwall TNTs supported Rh-nanoparticle catalysts would be very stable, based on our previous work, 37 and have a good catalytic activity for the hydroformylation of alkenes.
In the present contribution, we report multiwall TNTs supported Rh-nanoparticle catalysts prepared via three approaches and the synergy effects between nano-confine effect and size-dependent catalytic activity on the regioselective hydroformylation of vinyl acetate.
2. Experimental
2.1 Chemicals
All chemicals were purchased commercially and used without further purification. The gases, N2, CO and H2, used in the experiments were all of 99.99% purity.2.2 Synthesis of TNTs supported Rh-nanoparticle catalysts
The preparation procedures of TNTs supported Rh-nanoparticle catalysts are described as Scheme 2. All reduction processes were carried out under N2 atmosphere. | ||
| Scheme 2 Schematic processes of preparing Rh/TNTs. | ||
TNTs were synthesized by the hydrothermal treatment as the previous reports.32–36
The TNTs adsorbed Rh2+ ions were prepared via impregnation procedure using Rh2(Ac)4 as precursor. Typically, the synthesis was performed as follows: 1.0 g TNTs were dispersed in 20 mL of Rh2(Ac)4 aqueous solution (15.0 mmol L−1, 7.50 mmol L−1, 3.75 mmol L−1), and the system was vigorously agitated for 24 h. After low-energy sonication for 2 h, the mixture was centrifuged and green TNTs were obtained. In order to remove the Rh2(Ac)4 solution adsorbed on the outer surface of TNTs, the green TNTs were washed with a little distilled water.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Vwater = 9:1). Then, the mixture was refluxed, while vigorously stirred, for 6 h to reduce the Rh2+ ions adsorbed on TNTs. After cooling to room temperature, the mixture was centrifuged and the gray product was washed three times with ethanol. The product was dried at 40 °C for 12 h in vacuum.:Vwater = 9:1), and subsequently the mixture was irradiated for 4 h by using a 300 W high-pressure mercury lamp under stirring at ambient temperature. After irradiation, the obtained catalyst was centrifuged, washed with distilled water and ethanol. The product was dried at 40 °C for 12 h in vacuum.
:Vwater = 9:1). Then, the mixture was refluxed, while vigorously stirred, for 6 h to reduce the Rh2+ ions adsorbed on TNTs. After cooling to room temperature, the mixture was centrifuged and the gray product was washed three times with ethanol. The product was dried at 40 °C for 12 h in vacuum.:Vwater = 9:1), and subsequently the mixture was irradiated for 4 h by using a 300 W high-pressure mercury lamp under stirring at ambient temperature. After irradiation, the obtained catalyst was centrifuged, washed with distilled water and ethanol. The product was dried at 40 °C for 12 h in vacuum.2.3 Characterization of TNTs supported Rh-nanoparticle catalysts
The morphologies and microstructure of catalysts synthesized were analyzed by Transmission Electron Microscope (TEM, Philips T20ST). The chemical states of element Rh in catalysts were determined by X-ray Photoelectron Spectrometer (XPS, Kratos Axis Ultra DLD multi-technique X-ray photoelectron spectra), and all binding energies were calibrated using C 1s (Eb = 284.6 eV) as reference. The phase structures of catalysts were characterized with X-Ray Diffraction (XRD, Rigaku D/Max-2500 X-ray diffractometer with Cu Kα radiation). The bulk and surface Rh contents of samples were measured by Inductively Coupled Plasma (ICP-9000, USA Thermo Jarrell-Ash Corp) and EDX.2.4 Evaluation of catalytic performance of TNTs supported Rh-nanoparticle catalysts for hydroformylation
The catalytic hydroformylation reaction was carried out in a 250 mL stainless steel autoclave reactor equipped with a magnetic stirrer. In a typical experiment, the required amount of catalyst, substrate and solvent were added into the reactor. The reactor was sealed and purged three times with hydrogen, and subsequently pressurized to the desired pressure of H2 and CO. The reactor was heated up to reaction temperature and kept for desired time under stirring and at the controlled reaction temperature. After desired reaction time, the stirring was stopped, and the reactor was cooled down to room temperature, depressurized. The reaction mixtures were withdrawn for GC analysis (Shimadzu GC-2014 gas chromatograph equipped with a 30 m × 0.53 mm SE-30 capillary column and a FID).3. Results and discussion
3.1 Characterization of Rh/TNTs
First of all, the content and chemical state of the rhodium on the nanotubes were studied by ICP, EDX and XPS. EDX detection (Fig. 1) shows clearly that there are peaks of Rh, which confirms the presence of rhodium on the surface of TNTs. The EDX shows that the contents of Rh on the surfaces of Rh/TNTs-A, Rh/TNTs-B and Rh/TNTs-C are 0.79, 0.25 and 0.28 wt%, respectively, which are all higher than ICP results (Table 1). ICP and EDX results exhibit clearly Rh enrichment on the surfaces of samples prepared via different procedures, which is favorable for utilizing Rh effectively and catalytic reaction. | ||
| Fig. 1 EDX and XPS of Rh in Rh/TNTs-A (A), Rh/TNTs-B (B) and Rh/TNTs-C (C). | ||
| Entry | Catalyst | Rh content (wt%) | Conversion (%) | Aldehyde (%) | b:lb |
||
|---|---|---|---|---|---|---|---|
| ICP | EDX | XPSc (Rh0) | |||||
| a Reaction conditions: vinyl acetate = 5 mL, sub/Rh = 8000, catalyst = 0.40 g (7.0 × 10−3 mmol Rh), temp. = 100 °C, syngas pressure (CO/H2 = 1) = 4.0 MPa, solvent (toluene) = 65 mL, and reaction time = 8 h.b b:l is 2-acetoxy propanal:3-acetoxy propanal.c The ratios of Rh0 to total Rh in samples. |
|||||||
| 1 | Rh/TNTs-A | 0.19 | 0.79 | 85.94 | 22.98 | 68.91 | >99:1 |
| 2 | Rh/TNTs-B | 0.18 | 0.25 | 71.21 | 72.33 | 74.99 | 97:3 |
| 3 | Rh-TNTs-C | 0.18 | 0.28 | 87.35 | 99.37 | 72.90 | 98:2 |
Also displayed in Fig. 1 are Rh 3d XPS spectrums of Rh/TNTs-A, Rh/TNTs-B and Rh/TNTs-C. Binding energies of 307.2 and 312.0 eV are attributed to the Rh 3d5/2 and Rh 3d3/2, respectively, which are consistent with the literature data of Rh 3d5/2 and Rh 3d3/2 for the metallic Rh.39 The peaks centered at 309.3 eV and 314.1 eV, which are also attributed to the Rh 3d5/2 and Rh 3d3/2 species, clearly show the existence of Rh in oxidation state. It is well known that the low valent Rh is the active center for catalytic hydroformylation. The ratios of Rh0 to total Rh species in samples are calculated from the areas of the fitted Gaussian peaks and listed in Table 1.
The formation of Rh0 in the photoreducing procedure can be illustrated as eqn (1) and (2).35,36 TNTs absorb UV light and electron–hole pairs form Rh2+ ions capture the electrons from the conduction band of excited TiO2, and, as a consequence, Rh2+ ions become the metallic Rh.
| TiO2 + hv → TiO2 + e− + h+ | (1) |
| Rh2+ + 2e− → Rh0 | (2) |
The TEM images of the as-synthesized catalysts are shown in Fig. 2. As showed in Fig. 2, all of the catalysts take on perfect nanotubular morphology; their diameters are about 6 nm and lengths up to hundreds of nanometers. The Rh nanoparticles are well-dispersed on the surfaces of TNTs. By comparison with each other, the size of Rh particles in Rh/TNTs-A (Fig. 3-A) is the largest, the average size is about 3 nm. In the reducing process, the Rh atoms will be preferentially agglomerated on the just-formed Rh nuclei to generate multiple Rh islands on TNTs. This island growth mode can be attributed to the strong Rh–Rh interaction due to the high interatomic binding energy and surface free energy of Rh.40 When the strength of reductant is weak, the reduced Rh atoms have enough time to diffuse to other regions of TNT rather than agglomerating on the just-formed Rh nuclei. In the case of photoreducing process, though photogenerated electrons, the photoreductant, are strong reductant, their amount is not large enough to reduce Rh2+ ions rapidly on a large scale, which results in that Rh atoms is homogeneously and instantaneously produced during the irradiation thus providing favorable conditions for the formation of nearly monodispersed particles.41 However, compared with the photoreducing process, the concentration of strong reductant is far higher in the KBH4 reducing procedure, which reduces Rh2+ ions quickly and causes multiple Rh islands formation on the surfaces of TiO2 nanotubes. Compared with KBH4, ethanol is a weak reducing agent, and it is reasonable that Rh0 particles in Rh/TNTs-B are smaller than that in Rh/TNTs-A. It is now obvious that synthesizing size-tunable Rh0 particles can be achieved via the three methods, which is significant for preparing catalysts of size-dependent catalytic activity.
 | ||
| Fig. 2 The TEM images of the as-synthesized Rh/TNTs-A (A), Rh/TNTs-B (B), and Rh/TNTs-C (C). | ||
 | ||
| Fig. 3 XRD patterns of Rh/TNTs-A (A), Rh/TNTs-B (B) and Rh/TNTs-C (C). | ||
Fig. 3 shows the XRD patterns of Rh/TNTs-A, Rh/TNTs-B and Rh/TNTs-C. It can be seen from Fig. 3 that all of catalysts display the diffractions of anatase TiO2 (JCPDS21-1272); the Rh/TNTs-C has more perfect anatase phase, which is the result of UV irradiation.36 However, there is not any diffraction related to Rh0 particles due to the relatively low content and very small size of Rh particles.
3.2 Activity and selectivity of Rh/TNTs
To investigate the catalytic performances of Rh/TNTs-A, Rh/TNTs-B and Rh/TNTs-C, hydroformylation of vinyl acetate was chosen as a model reaction so that both catalytic activity and regioselectivity of catalysts could be evaluated in detail; the results are listed in Table 1. Of the three catalysts, though their bulk Rh contents are almost identical, the Rh/TNTs-A has the lowest activity, which is a typical size-dependent catalytic property. As discussed above, the size of Rh0 particles in Rh/TNTs-A is the largest, which results in that the amount of catalytically Rh active species is small in spite of the fact that the percentage of Rh0 in Rh/TNTs-A is not the lowest. However, for both Rh/TNTs-B and Rh/TNTs-C, though the difference in Rh nanoparticle size is not obvious, Rh/TNTs-C shows higher activity than Rh/TNTs-B. The reasonable interpretation is that the Rh/TNTs-C has a higher percentage of Rh0, which would be more favorable for coordination of reactants to the metallic Rh, resulting in good activity. The leaching of transition-metal nanoparticle catalyst is frequent, especially for the catalytically active group VIII metals. Therefore, the Rh leaching of Rh/TNTs-C in hydroformylation of vinyl acetate was studied. The leaching of Rh in solution was detected by ICP-AES and the results indicated that the leaching of Rh was 1.013 ppm (0.071 mg), which is the 10 percent of Rh species in Rh/TNTs-C. According to the data of XPS, the ratio of Rh2+ to total Rh species in Rh/TNTs-C is 12.65%, indicating that the leaching of Rh species is merely the TNTs adsorbed Rh2+ and Rh nanoparticles on TNTs are stable in the hydroformylation. This result is in accordance with our previous work of Co–B/TNTs for hydroformylation of cyclohexene.37Shown in Table 2 are the catalytic performances of Rh/TNTs-C catalysts with various Rh loadings. It can be seen that increasing Rh loading can remarkably enhance the conversion of vinyl acetate and TOF, but the selectivity for aldehyde and regioselectivity decrease slightly with the increase of Rh loading from 0.07 wt% to 0.18 wt%. The optimum Rh loading should be about 0.18 wt%.
| Entry | Rh content (wt%) | Conversion (%) | TOFb (h−1) | Aldehyde (%) | b:lc |
|---|---|---|---|---|---|
| a Reaction conditions: vinyl acetate = 5 mL, Rh/TNTs-C = 0.40 g, temp. = 100 °C, syngas pressure (CO/H2 = 1) = 6.0 MPa, solvent (toluene) = 65 mL, and reaction time = 3 h.b TOF = number of moles of product formed/(number of moles of Rh × h).c b:l is 2-acetoxy propanal:3-acetoxy propanal. |
|||||
| 1 | 0.07 (2.7 × 10−3 mmol) | 30.33 | 2080 | 83.62 | >99:1 |
| 2 | 0.09 (3.5 × 10−3 mmol) | 39.74 | 2120 | 74.74 | >99:1 |
| 3 | 0.18 (7.0 × 10−3 mmol) | 100 | 2670 | 74.70 | 95:5 |
For the heterogeneous catalysis catalyzed liquid-phase hydroformylation reaction, the influence of reaction medium should be a crucial factor to consider. Listed in Table 3 are catalytic performances of Rh/TNTs-C in toluene, isooctane, tetrahydrofuran, ethanol and neat vinyl acetate without solvent. As the results showed, Rh/TNTs-C displays higher catalytic activity in solvents of weaker Lewis basicity (Table 3, entries 1 and 2) than in coordinative solvents (Table 3, entries 3 and 5); the regioselectivities are almost identical in two types of solvents. The results are very reasonable. It is well accepted that coordination of reactants to the catalytically active species is the first and a necessary step for catalytic hydroformylation, the catalytic activity, rather than selectivity, of catalyst is certainly affected when catalytically active centers is occupied by coordinative solvent. It is noteworthy that the conversion of vinyl acetate is 78.96%, but the main production is acetal and the selectivity for acetal is 69.69% when ethanol is used as solvent (Table 3, entry 4). For neat vinyl acetate without solvent in the reaction system (Table 3, entry 5), the conversion of vinyl acetate decreases to 19.50%, which is due to the increase of sub/Rh from 8 × 103 to 112 × 103, and the selectivity for aldehyde is still maintained with good regioselectivity for 2-acetoxy propanal (2-acetoxy propanal:3-acetoxy propanal is 99:1).
| Entry | Solvent | Conversion (%) | Aldehyde (%) | Acetal (%) | b:lb |
|---|---|---|---|---|---|
| a Reaction conditions: vinyl acetate = 5 mL, sub/Rh = 8000, Rh/TNTs-C = 0.40 g (7.0 × 10−3 mmol Rh), temp. = 100 °C, syngas pressure (CO/H2 = 1) = 6.0 MPa, solvent = 65 mL, and reaction time = 3 h.b b:l is 2-acetoxy propanal:3-acetoxy propanal.c No solvent, vinyl acetate = 70 mL. |
|||||
| 1 | Toluene | 100 | 74.70 | 0 | 95:5 |
| 2 | Isooctane | 96.53 | 79.60 | 0 | 95:5 |
| 3 | Tetrahydrofuran | 33.94 | 77.90 | 0 | 95:5 |
| 4 | Ethanol | 78.96 | 26.51 | 69.69 | 95:5 |
| 5c | — | 19.50 | 74.81 | 0 | 99:1 |
Besides reaction medium and Rh loading, the reaction temperature and pressure may also influence reaction rate and selectivity. The effects of reaction temperature and CO/H2 pressure on the hydroformylation are examined in detail and the results are summarized in Table 4. In the pressure range of 4.0–6.0 MPa (Table 4, entries 1–3), the conversion of vinyl acetate increases with the increase of the syngas pressure, while the regioselectivities for 2-acetoxy propanal decreases slightly at the same reaction temperature. As showed in Table 4, the reaction temperature also affects conversion and regioselectivity of reaction. With the increase of temperature from 60 °C up to 100 °C, the conversion of vinyl acetate increases along with decrease in regioselectivity for 2-acetoxy propanal. At lower reaction temperature (60 °C), high selectivity for aldehyde is observed (91.49%), however the TOF of vinyl acetate is rather low (890 h−1). At 100 °C, the conversion of vinyl acetate increases up to 100% with TOF increased to 2670 h−1 and selectivity for aldehyde decreased to 74.70%.
| Entry | Pressure (MPa) | Temperature (°C) | Conversion (%) | TOFc (h−1) | Aldehyde (%) | b:ld |
|---|---|---|---|---|---|---|
| a Reaction conditions: vinyl acetate = 5 mL, sub/Rh = 8000, Rh/TNTs-C = 0.40 g (containing 7.0 × 10−3 mmol Rh), CO/H2 = 1, solvent (toluene) = 65 mL, and reaction time = 3 h.b Reaction conditions: vinyl acetate = 5 mL, sub/Rh = 8000, Rh/TNTs-C = 0.40 g (containing 7.0 × 10−3 mmol Rh), syngas pressure (1:1), solvent (toluene) = 65 mL, and reaction time = 6 h.c TOF = number of moles of product formed/(number of moles of Rh × h).d b:l is 2-acetoxy propanal:3-acetoxy propanal. |
||||||
| 1 | 4.0 | 100 | 75.08 | 2000 | 77.84 | 99:1 |
| 2 | 5.0 | 100 | 88.89 | 2370 | 75.21 | 98:2 |
| 3 | 6.0 | 100 | 100 | 2670 | 74.70 | 95:5 |
| 4b | 6.0 | 60 | 66.87 | 890 | 91.49 | >99:1 |
| 5b | 6.0 | 80 | 99.92 | 1330 | 86.23 | 97:3 |
Fig. 4 shows the variations of conversion, selectivity for aldehyde and 2-acetoxy propanal/3-acetoxy propanal ratio with reaction time. It is can be seen from the data in Fig. 4 that prolonging reaction time led to an increase in conversion. Before 1 h, the conversion of vinyl acetate increases quickly with prolonging reaction time; almost all of vinyl acetate is consumed after 2 h. The chemoselectivity for aldehyde and regioselectivity for 2-acetoxy propanal keep nearly unchangeable in the catalytic process for 4 h, which means that the Rh/TNTs-C has high regioselectivity for 2-acetoxy propanal.
 | ||
| Fig. 4 Variations of conversion, chemoselectivity and regioselectivity with reaction time. | ||
As discussed above, the high regioselectivity (2-acetoxy propanal:3-acetoxy propanal > 95:5) is an unchanged result in Rh/TNTs catalyzed vinyl acetate hydroformylation.
3.3 Mechanism
On the basis of experimental results and reported literature1,2,42–44 on vinyl acetate hydroformylation, the proposed reaction procedures of vinyl acetate hydroformylation over Rh/TNTs catalysts is shown in Scheme 3. Firstly, the Rh particles turn into RhHy(CO)x/TNTs in the atmosphere of H2 and CO. The catalytic cycle involves successively the coordination of the vinyl acetate, the formation of intermediate (I) or (II), and so on; furthermore, the formation of (I) or (II) dominantly decides the regioselectivity of the reaction for 2-acetoxy propanal and 3-acetoxy propanal. It is well understandable that the formation of (I) and (II) is related to the inductive effect and chelating effect of the ester carbonyl group, as well as the relative stability of (I) and (II). It has been reported that (I) with a five-membered ring is a more stable intermediate45 and the next intermediate is a stable six-membered ring, which should be the reason that the dominant 2-acetoxy propanal:3-acetoxy propanal ratio (>9:1) comes into being in the catalytic hydroformylation of vinyl acetate over Rh/TNTs.
 | ||
| Scheme 3 Proposed mechanism for Rh/TNTs catalyzed hydroformylation of vinyl acetate. | ||
4. Conclusion
(1) The TNTs supported Rh-nanoparticle catalysts prepared via three methods show the perfect nanotubular morphology, and Rh particles are well-dispersed on the surfaces of TNTs though their sizes are different.(2) The size distribution of TNTs supported Rh nanoparticles is dependent on the reducing agent used, which plays the important role from the view of catalytic activity.
(3) The catalytic activities of these catalysts in solvents of weaker Lewis basicity are higher than that in coordinative solvents.
(4) Though all these catalysts display good catalytic performances and high regioselectivity for 2-acetoxy propanal in the hydroformylation reaction of vinyl acetate, Rh/TNTs-C catalyst prepared by impregnation-photoreducing procedure shows the highest catalytic activity.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (21373120, 21301098, 21071086 and 21271110), the Applied Basic Research Programs of Science and Technology Commission Foundation of Tianjin (13JCQNJC02000 and 12JCYBJC13100) and project IRT13022, B12015.References
- A. A. Dabbawala, H. C. Bajaj, G. V. S. Rao and S. H. R. Abdi, Appl. Catal., A, 2012, 419–420, 185 CrossRef CAS PubMed.
- A. A. Dabbawala, R. V. Jasra and H. C. Bajaj, Catal. Commun., 2010, 11, 616 CrossRef CAS PubMed.
- D. B. G. Williams, M. Ajam and A. Ranwell, Organometallics, 2007, 26, 4692 CrossRef CAS.
- G. M. Noonan, D. Newton, C. J. Cobley, A. Suárez, A. Pizzano and M. L. Clarke, Adv. Synth. Catal., 2010, 352, 1047 CrossRef CAS.
- C. X. Cai, S. C. Yu, B. N. Cao and X. M. Zhang, Chem.–Eur. J., 2012, 18, 9992 CrossRef CAS PubMed.
- D. Selent, R. Franke, C. Kubis, A. Spannenberg, W. Baumann, B. Kreidler and A. Börner, Organometallics, 2011, 30, 4509 CrossRef CAS.
- S. Yu, Y. Chie and X. Zhang, Adv. Synth. Catal., 2009, 351, 537 CrossRef CAS.
- O. Saidi, J. Ruan, D. Vinci, X. Wu and J. Xiao, Tetrahedron Lett., 2008, 49, 3516 CrossRef CAS PubMed.
- N. Sudheesh, A. K. Chaturvedi and R. S. Shukla, Appl. Catal., A, 2011, 409–410, 99 CrossRef CAS PubMed.
- R. Jennerjahn, I. Piras, R. Jackstell, R. Franke, K. Wiese and M. Beller, Chem.–Eur. J., 2009, 15, 6383 CrossRef CAS PubMed.
- W. Alsalahi and A. M. Trzeciak, RSC Adv., 2014, 4, 30384 RSC.
- I. Piras, R. Jennerjahn, R. Jackstell, A. Spannenberg, R. Franke and M. Beller, Angew. Chem., 2011, 123, 294 CrossRef.
- Y. Yuki, K. Takahashi, Y. Tanaka and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 17393 CrossRef CAS PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. EI-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS PubMed.
- I. Nakamula, Y. Yamanoi, T. Imaoka, K. Yamamoto and H. Nishihara, Angew. Chem., Int. Ed., 2011, 50, 5830 CrossRef CAS PubMed.
- A. Gniewek and A. M. Trzeciak, Top. Catal., 2013, 56, 1239 CrossRef CAS PubMed.
- X. Y. Quek, Y. J. Guan and E. J. M. Hensen, Catal. Today, 2012, 183, 72 CrossRef CAS PubMed.
- T. Takanashi, M. Tamura, Y. Nakagawa and K. Tomishige, RSC Adv., 2014, 4, 28664 RSC.
- S. Agarwal and J. N. Ganguli, J. Mol. Catal. A: Chem., 2013, 2, 44 CrossRef PubMed.
- S. Akbayrak, S. Gençtürk, İ. Morkan and S. Özkar, RSC Adv., 2014, 4, 13742 RSC.
- J. A. Dahl, B. L. S. Maddux and J. E. Hutchison, Chem. Rev., 2007, 107, 2228 CrossRef CAS PubMed.
- M. Boutros, G. Shirley, T. Onfroy and F. Launay, Appl. Catal., A, 2011, 394, 158 CrossRef CAS PubMed.
- Z. Sun, Y. H. Wang, M. M. Niu, H. Q. Yi, J. Y. Jiang and Z. L. Jin, Catal. Commun., 2012, 27, 78 CrossRef CAS PubMed.
- B. R. Sathe, RSC Adv., 2012, 2, 3735 RSC.
- R. Narayanan and M. A. El-Sayed, Chim. Oggi, 2007, 25, 84 CAS.
- C. A. Stowell and B. A. Korgel, Nano Lett., 2005, 5, 1203 CrossRef CAS.
- M. E. Grass, S. H. Joo, Y. W. Zhang and G. A. Somorjai, J. Phys. Chem. C, 2009, 113, 8616 CAS.
- Y. Borodko, S. E. Habas, M. Koebel, P. D. Yang, H. Frei and G. A. Somorjai, J. Phys. Chem. B, 2006, 110, 23052 CrossRef CAS PubMed.
- Y. Borodko, S. M. Humphrey, T. D. Tilley, H. Frei and G. A. Somorjai, J. Phys. Chem. C, 2007, 111, 6288 CAS.
- D. F. Han, X. H. Li, H. D. Zhang, Z. M. Liu, G. S. Hu and C. Li, J. Mol. Catal. A: Chem., 2008, 283, 15 CrossRef CAS PubMed.
- D. F. Han, X. H. Li, H. D. Zhang, Z. M. Liu, J. Li and C. Li, J. Catal., 2006, 243, 318 CrossRef CAS PubMed.
- J. J. Song, B. L. Zhu, W. L. Zhao, X. J. Hu, Y. K. Shi and W. P. Huang, J. Nanopart. Res., 2013, 15, 1494 CrossRef.
- H. Q. An, J. X. Li, J. Zhou, K. R. Li, B. L. Zhu and W. P. Huang, J. Mater. Chem., 2010, 20, 603 RSC.
- H. Q. An, B. L. Zhu, J. X. Li, J. Zhou, S. R. Wang, S. M. Zhang, S. H. Wu and W. P. Huang, J. Phys. Chem. C, 2008, 112, 18772 CAS.
- H. Q. An, J. Zhou, J. X. Li, B. L. Zhu, S. R. Wang, S. M. Zhang, S. H. Wu and W. P. Huang, Catal. Commun., 2009, 11, 175 CrossRef CAS PubMed.
- B. L. Zhu, K. R. Li, J. Zhou, S. R. Wang, S. M. Zhang, S. H. Wu and W. P. Huang, Catal. Commun., 2008, 9, 2323 CrossRef CAS PubMed.
- X. J. Hu, Y. K. Shi, Y. J. Zhang, B. L. Zhu, S. M. Zhang and W. P. Huang, Catal. Commun., 2015, 59, 45 CrossRef CAS PubMed.
- N. Liu, X. Y. Chen, J. L. Zhang and J. W. Schwanka, Catal. Today, 2014, 225, 34 CrossRef CAS PubMed.
- G. Pótári, D. Madarász, L. Nagy, B. László, A. Sápi, A. Oszkó, A. Kukovecz, A. Erdőhelyi, Z. Kónya and J. Kiss, Langmuir, 2013, 29, 3061 CrossRef PubMed.
- S. F. Xie, H. C. Peng, N. Lu, J. G. Wang, M. J. Kim, Z. X. Xie and Y. N. Xia, J. Am. Chem. Soc., 2013, 135, 16658 CrossRef CAS PubMed.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS PubMed.
- N. Sivasankar and H. Frei, J. Phys. Chem. C, 2011, 115, 7545 CAS.
- S. S. C. Chuang and F. Guzman, Top. Catal., 2009, 52, 1448 CrossRef CAS.
- Y. Zhang, K. Nagasaka, X. Q. Qiu and N. Tsubaki, Appl. Catal., A, 2004, 276, 103 CrossRef CAS PubMed.
- Y. L. Borole and R. V. Chaudhari, Ind. Eng. Chem. Res., 2005, 44, 9601 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11156g |
| This journal is © The Royal Society of Chemistry 2014 |