
Rapid multilayer construction on a non-planar substrate by layer-by-layer self-assembly under high gravity†
Abstract
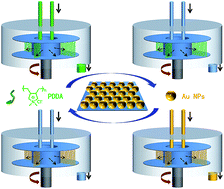
Rapid construction of layer-by-layer (LbL) self-assembled multilayers on non-planar substrates is challenging because most conventional LbL processes are time consuming, which restricts further applications of LbL in industry and its commercialization. Therefore, herein we introduced the high gravity (HG) technique, which is a well-established industrial chemical engineering process for intensification of mass transfer, into the LbL assembly process to realize rapid film deposition on porous nickel foam. By using a model system of electrostatically driven PDDA/AuNPs multilayers, the adsorption kinetics, LbL procedure and film morphology have been examined under both conventional dipping conditions and a HG field. The results show that the time to reach saturated adsorption of building blocks with the HG field has been shortened remarkably by up to 16 times while the film quality remains identical. In this way, the fabrication of LbL multilayers can be highly accelerated in the presence of a HG field without disturbing the film quality on non-planar substrates. Moreover, the mechanism for the rapid construction of LbL multilayers using the HG technique is interpreted using the boundary layer theory that the highly turbulent flow in the HG field enhanced the mass transfer rate for the rapid adsorption of building blocks onto substrates.
 Please wait while we load your content...
Please wait while we load your content...

