
(S)-5-Benzyl- and 5-benzylidene-imidazo-4-one derivatives synthesized and studied for an understanding of their thermal reactivity†
Abstract
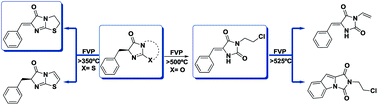
Flash vacuum as well as static pyrolysis were used to gain insight into the mechanisms of decomposition of the title compounds. They were synthesized by the use of microwave techniques that allowed better yields than the established methods. Since the benzyl compounds always open a radical channel in the thermal processes, consequently lowering the yields of other important intramolecular processes, we also started the pyrolysis with benzylidene derivatives. Detection and quantification of products accompanied by kinetic analyses were carried out for both types of compounds. The activation energies as well as the entropy contributions have been determined. Moreover, DFT calculations provided support for and corroborating of the proposed thermal pathways.
 Please wait while we load your content...
Please wait while we load your content...

