
Twin applications of highly selective Cu2+ fluorescent chemosensor and cytotoxicity of 2-(2-phenylhydrazono)-1H-indene-1,3(2H)-dione and 2-(2-(4-methoxyphenyl)hydrazono)-1H-indene-1,3(2H)-dione: molecular docking and DFT studies†
Annamalai Subhasria,
Chinnadurai Anbuselvan*a and
Nagarajen Rajendraprasadb
aDepartment of Chemistry, Annamalai University, Annamalainagar-608 002, India. E-mail: cas_amu@yahoo.co.in
bDepartment of Biochemistry and Biotechnology, Annamalai University, Annamalai Nagar-608 002, India
First published on 23rd October 2014
Abstract
Fluorescent chemosensors based on 2-(2-phenylhydrazono)-1H-indene-1,3(2H)-dione (1) and 2-(2-(4-methoxyphenyl)hydrazono)-1H-indene-1,3(2H)-dione (2) with high sensitivity and selectivity toward paramagnetic Cu2+ were developed over other cations. The Cu2+-induced fluorescence turn-on mechanism was revealed to be mediated by intramolecular charge transfer from the ligand to the metal. The compounds were characterized by UV-vis, FT-IR, 1H and 13C NMR techniques, scanning electron microscopy (SEM), energy dispersive spectroscopy (EDS), elemental color mapping and fluorescence spectroscopy. Morphology changes of compounds (1) and (2) to complex with Cu2+ were also investigated by SEM. EDS analysis exposed the occurrence of copper in complexes. Elemental color mapping also supported the copper present in the complexes of corresponding compounds (1) and (2). The metal sensing (chemosensing) properties of compounds (1) and (2) were also examined via fluorescence spectroscopy. The sensor showed excellent selectivity with fluorescence enhancement to copper over other cations in ethanolic solution. The chelating functionality of compounds (1) and (2) was evaluated for inhibitory properties against various human cancer proteins like 4LRH, 4EKD, 4GIW and 4L9K using an online docking server. The cytotoxicity study demonstrated the ability of compounds (1) and (2) to inhibit the growth of KB cell lines. Combined experimental and theoretical studies were carried out on the molecular structure using density functional methods (B3LYP) invoking the 6-31G basis set. The density functional theory (DFT) calculation was carried out for both compounds. Energy of the highest occupied molecular (HOMO) orbital and lowest unoccupied (LUMO) molecular orbitals were predicted.
Introduction
An improvement in expedient selective fluorescent chemosensors for identifying important species, such as certain transition, non-transition metal ions and anions, is of current interest because of various biological and environmental difficulties that cause serious problems for human health and ecological systems. Chemical sensing refers to the continuous monitoring for the presence of chemical species.1–5 Many disciplines require sensing systems, including chemistry, biology, clinical biology and environmental science.Expensive and complicated methods, such as atomic absorption,6 inductively coupled plasma, and atomic emission spectroscopy7 have been employed in detection of toxic metal ions. Fluorescent chemosensors are often used to detect many ions due to their simplicity and sensitivity, low cost, high selectivity and quick response.8,9 The classical design of a fluorescent indicator includes two moieties, a receptor responsible for the molecular recognition of the analyte and a fluorophore responsible of signaling the recognition event. Copper is a vital element in the human body, a co-factor in various enzymes and copper-based pigments. Despite its significant roles in organisms, the accumulation of excess amounts of copper ions or their misregulation can cause a number of cruel diseases. It is believed that the disruption of copper homeostasis is implicated in certain neurodegenerative disorders, including Alzheimer's and Parkinson's diseases.10,11 Interest in the design and synthesis of fluorescent chemosensors with a Cu2+-induced “turn-on” fluorescence signal has increased in recent years. However, only a few sensors are currently available because implementation of sensing probes in functional devices without loss of sensitivity remains a major challenge.12 Lu et al. have reported a functional oligonucleotide-based “turn-on” fluorescent probe for detection of Cu2+ in aqueous solution with satisfying sensitivity and selectivity.13 Swamy et al. have proposed a monoboronic acid-conjugated, rhodamine-based probe for Cu2+ with reversible fluorescence “turn-on” response and high selectivity, and they successfully applied it to image Cu2+ in living cells and organisms.14
Aryl hydrazones possess desirable biological and pharmacological properties, such as antibacterial, antiviral, antineoplastic, and antimalarial behaviors.15 The resonance-assisted hydrogen bond systems engage a synergistic reinforcement of hydrogen bonds by delocalization of a p-conjugated chain connecting donor and acceptor atoms; they have been applied for activation of a carbon in α-position to a carbonyl, induced enolization in keto–enol tautomerism, controlled crystal packing, formation of H-bonds in functional molecular materials, activation of dinitriles towards formation of amidines, carboxamides and iminoesters, and so on. Special attention should be focused on the nature of the strong intramolecular O⋯H–N resonance-assisted hydrogen bond and its influence on the enol–azo hydrazone transformation.16–19 The rich tautomerism and isomerism of aryl hydrazones together with the intramolecular resonance-assisted hydrogen bond system can be applied for regulation of tautomerization–isomerization; activation of the carbon in α-position to a carbonyl, antiferroelectric paraelectric transition, regioselective activation of dinitriles, catalysis, ligand liberation, and so on.20 In some cases hydrogen bonding acts as an active site for initiation of chemical reactions. These results led to the observation that the system exists in a conformation with a preferred orientation where the stereoelectronic constraints and stabilizing effect of hydrogen bonding are competing.
In this article, we demonstrate the use of compounds (1) and (2) as a highly efficient fluorescent chemosensor for Cu2+. In terms of sensitivity concerns, chemosensors exhibiting fluorescence enhancement (fluorescence “turn-on”) upon Cu2+ ion complexation are favoured. Among these detection approaches for the Cu2+ ion, fluorescence spectroscopy was used because of its high sensitivity, high selectivity and low cost. Our sensor shows simple and good selectivity compared to recently developed Cu2+ sensors21–28 attributed to the very high association constants for binding Cu2+.
Experimental
All solvents were analytical reagent grade. The homogeneity of the compounds was monitored by ascending thin-layer chromatography (TLC) on silica gel-G (Merck) coated aluminum plates, visualized by iodine vapor and UV light. IR spectra were recorded on an Avatar Nicholet FT-IR spectrophotometer (range 4000–400 cm−1) in KBr pellets (λmax in cm−1). 1H and 13C NMR spectra for analytical purposes were recorded in CDCl3 on a Bruker instrument at 400 MHz, chemical shifts were expressed in δ-scale downfield from TMS as an internal standard. SEM analysis was performed for surface morphology of compound (1), compound (2) and corresponding complexes with Cu2+, on platinum-coated samples using a JEOL JSM-5610 SEM. EDS analysis was performed upon using a JEOL JSM-5610 SEM equipped with EDS. UV-vis spectra were recorded on a Shimadzu UV-1650PC spectrophotometer by dissolving the sample in spectral grade ethanol using a 1 cm path-length quartz cell. A PerkinElmer LS 55 fluorescence spectrometer was employed to record the fluorescence (FL) spectra at room temperature. The choice of excitation wavelengths was based on absorbance spectral characteristics.Preparation of compounds
Compounds were prepared by the general procedure (Scheme 1) in which anilines (0.5 mmol) were dissolved separately in 1 N HCl (25 cm3) at 0–5 °C, and in each case cooled aqueous solution (10 cm3) of NaNO2 (0.40 g) was added drop-wise with stirring followed by the addition of indene-1,3-dione (0.73 g, 0.5 mmol) and sodium acetate (5.0 g) dissolved in water (30 cm3). Corresponding mixtures were further stirred for 4 h at room temperature (25 °C). Solids thus obtained were filtered and washed several times with water followed by ethanol and then dried in vacuum. The crude products were recrystallized in ethanol. Compounds (1) and (2) were purified by column chromatography by using benzene as the eluent. Yield and melting points of the derived compounds are mentioned below. | ||
| Scheme 1 Schematic representation of hydrozone synthesis. | ||
Computational details
Theoretical investigations of unbound compounds (1), (2) and bound with Cu2+ were performed with Gaussian 03W programme package,29 invoking gradient geometry optimization at the B3LYP level, and the LANL2DZ effective core potential was used for all atoms.30 Initial geometry generated from standard geometrical parameters was minimized without any constraints in the potential energy surface at ab initio, adopting the standard 6-31G basis set.Results and discussion
2-(2-Phenyl)hydrazono-2H-indene-1,3-dione (1)
Yellow solid, yield: 92%, m. p. 197 °C; IR (KBr, cm−1) 3421, 3083, 3043, 3018, 2922, 2850, 1718, 1664, 1539 (Fig. S1†); 1H NMR (400 MHz, CDCl3) (δ): 7.22–7.96 (aromatic protons), 13.48 (s, NH) (Fig. S3†); 13C NMR (125 MHz, CDCl3) (δ) 116.3, 122.9, 123.2, 125.7, 126.3, 127.3, 128.3, 129.7, 135.0, 135.3, 140.6, 141.0, 144.1, 186.1, 188.0 (Fig. S4†).2-(2-(4-Methoxyphenyl)hydrazono)-2H-indene-1,3-dione (2)
Orange solid, yield: 87%, m. p. 190 °C; IR (KBr, cm−1) 3431, 3154, 2962, 2923, 2851, 1711, 1671, 1592 (Fig. S2†); 1H NMR (400 MHz, CDCl3) (δ): 3.85 (s, OCH3), 6.95–7.93 (aromatic protons), 13.63 (s, NH) (Fig. S5†); 13C NMR (125 MHz, CDCl3) (δ) 55.7, 115.0, 117.8, 122.6, 123.0, 129.9, 134.5, 134.7, 135.0, 138.7, 140.4, 158.5, 186.4, 188.9 (Fig. S6†).The arylhydrazones of 2-(2-phenyl)hydrazono-2H-indene-1,3-dione (1) and 2-(2-(4-methoxyphenyl)hydrazono)-2H-indene-1,3-dione (2) have been synthesized and their IR spectra for 1 and 2 show ν(NH) vibration at 3421–3431 cm−1, while ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ν(CO⋯H) and ν(CN) are observed at 1718–1711, 1664–1671 and 1539–1592 cm−1, respectively (Fig. S1 and S2†). These bands can be related to the intramolecular hydrogen-bonded hydrazone fragment. This conclusion was further supported by the 1H and 13C NMR spectrum; the 1H NMR spectra of (1) and (2) showed signals at 13.48–13.63 ppm, which can be assigned to the proton of the NH moiety adjacent to the aryl unit. In compound 2 the methoxy proton appeared at 3.85 ppm. In compound 1 and 2, the aromatic proton signals appear in the downfield region of 6.95–7.96 ppm with the expected splitting patterns. Carbonyl sp2 carbon atoms appear as separate signals in the low field region of about 186 and 188 ppm for C-(1) and C-(2), respectively. Strong intramolecular N–H⋯O–C hydrogen bonding deshields C-(1) with respect to C-(2) to the extent of about 2 ppm.
O), ν(CO⋯H) and ν(CN) are observed at 1718–1711, 1664–1671 and 1539–1592 cm−1, respectively (Fig. S1 and S2†). These bands can be related to the intramolecular hydrogen-bonded hydrazone fragment. This conclusion was further supported by the 1H and 13C NMR spectrum; the 1H NMR spectra of (1) and (2) showed signals at 13.48–13.63 ppm, which can be assigned to the proton of the NH moiety adjacent to the aryl unit. In compound 2 the methoxy proton appeared at 3.85 ppm. In compound 1 and 2, the aromatic proton signals appear in the downfield region of 6.95–7.96 ppm with the expected splitting patterns. Carbonyl sp2 carbon atoms appear as separate signals in the low field region of about 186 and 188 ppm for C-(1) and C-(2), respectively. Strong intramolecular N–H⋯O–C hydrogen bonding deshields C-(1) with respect to C-(2) to the extent of about 2 ppm.
Solvatochromism
The absorption spectra of compounds (1) and (2) were measured in various solvents of different polarity and shown in Fig. S7A and B.† The longest wavelength absorption band has been assigned to n–π* transition. This band is associated with a charge transfer from the N–H group to the carbonyl oxygen (CO) due to intramolecular hydrogen bonding between them. This intramolecular hydrogen bond is responsible for stabilizing the charge transfer band. A shoulder at a shorter wavelength around 480 nm is due to the π–π* transition. In particular, for hydazone derivatives charge transfer between a donor part, N–H (D) and an acceptor part, indene-1,3-dione (CO), has been considered. Both donor and acceptor appear to be localized in the quasi-aromatic rings (Fig. 9) containing the intramolecular hydrogen bonds. Thus, these hydrogen-bonded, quasi-aromatic rings contribute to stabilization of the charge transfer band.
The emission spectra of compounds (1) and (2) were studied in various solvents. The results are summarized in Table S1† and the spectrum of compounds (1) and (2) in various solvents are depicted in Fig. S8A and B.† Compound (1) showed two emission bands: one band occurred at around 460 nm and another band at around 480 nm. It is assumed that the longer one pertains to charge transfer (CT) and the shorter one to local excitation (LE).31–33 In both compounds, the photoinduced electron transfer (PET) from donor to acceptor produces a low-lying anomalous CT state that leads to new longer wavelength fluorescence. The emission maximum for both states was found to be independent of the excitation wavelength. In protic solvents, protonation of the fluorophore increased the oxidation state of the donor group, which resulted in the LE of fluorescence only. The LE state was more enhanced in intensity when compared to the CT state due to the increased charge density on nitrogen.
Similarly, compound (2) had the LE at 485 nm and the CT band at 515 nm. The variations in solvatochromic properties of compounds (1) and (2) depend on the substitution pattern. The red shift of the emission maximum increases in –OCH3 substituted compound (2) when compared to compound (1). This observation reveals the significant influence of H-bonding on the solvatochromic properties, presumably as a result of stabilization of the negative charge at the carbonyl oxygen atoms during solvent relaxation.
To better understand the solvent polarity effect, the Lippert–Mataga relation was applied. This relation has been widely used to correlate the energy difference between absorption (νa) and emission (νf), also known as the Stokes shift, with solvent polarity represented by Δf. This relation, given in eqn (1), involves both the dielectric constant and the refractive index (n) of the solvents:34,35
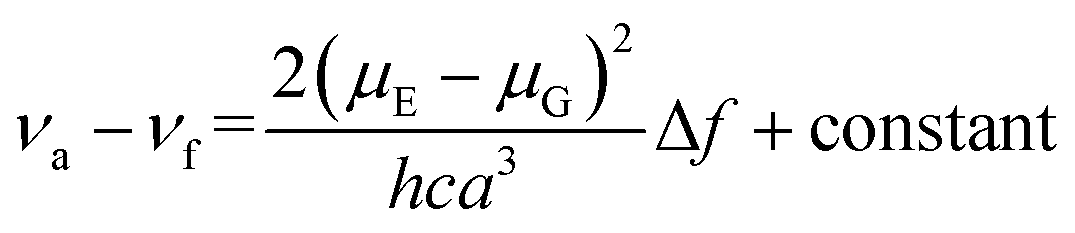 | (1) |
In eqn (1), νa and νf are the wavenumbers (cm−1) corresponding to absorption and emission, respectively; h is Planck's constant, c is the speed of light; and a is the radius of the solvent cavity in which the fluorophore resides. For an elongated molecule shape, a is usually estimated as 40% of its longest axis.36 The term (Δf) involving ε and n is called the orientation polarizability, which only accounts for spectral shifts due to reorientation of the solvent molecules. Therefore, the Lippert–Mataga relation is based on the assumption that the energy difference is only proportional to the solvent orientation polarizability (known as the general solvent effect). Inability of the Stokes shift to increase linearly with Δf usually implies that specific solvent effects are involved.
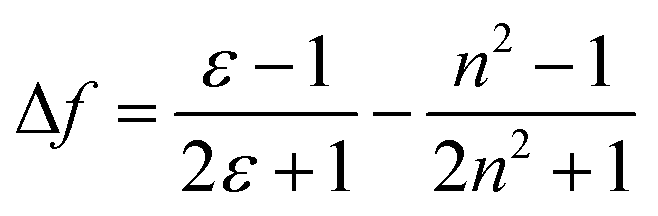 | (2) |
Fig. S9† shows the Lippert–Mataga plots of compounds (1) and (2) in 13 organic solvents. The estimation from the Lippert–Mataga equation is based on the assumption that the photophysical properties of compounds (1) and (2) can be described by the general solvent effect; hence, it may not hold if specific solvent effects are involved. The Stokes shift slightly increases with increasing polarity of the solvent (as expected, based on the CT character of the excited state) and does not follow linear dependence on f(ε, n) in all solvents used, that is, solvent-specific interactions were observed. The linear variation of the Stokes shift with ET (30) (with the solvent polarity parameter) is shown in Fig. S10.† The compounds (1) and (2) exhibited a slight increase or no increase in Stokes shift from non-polar to polar aprotic solvents, mainly due to the combined effect of increasing polarity of the medium and intramolecular charge transfer (CT) state, and it confirmed that solvent polarity cannot affect the Stokes shift of compounds (1) and (2). The νa − νf value is unusually small in hexane, and remarkably, deviates from the linear correlation. This deviation was particularly connected with the decrease of λF in hexane and indicated a different charge redistribution of the excited state of 1 than in other solvents. This suggested that the solvent polarity might not be the only factor affecting the spectral shifts. Specific solvent effects including hydrogen bonding, acid–base chemistry, and charge transfer interactions can also result in nonlinear Lippert–Mataga plots.
Chemosensor
The response of compounds (1) and (2) to various metal ions were investigated in aqueous buffer. A very distinctive selectivity for copper ions was displayed, as observed via fluorescence spectroscopy. When compounds (1) and (2) were treated with various metal ions, such as Cd2+, Ni2+, Pb2+, Zn2+, Ag+, Hg2+, Cu2+, Mn2+, Fe2+, Cr3+, K+, Fe2+, Fe3+, Al3+ and Co2+ in the buffer, complete fluorescence enhancement was observed only for Cu2+ ions. The fluorescence responses of the sensors (1) and (2) to various cations and its selectivity for Cu2+ are illustrated in Fig. 6A and 7A. The fluoroionophoric behavior of sensors (1) and (2) were investigated in ethanol–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution [sensor (1) and (2)] = 1 and 2 μM, [Cu2+] = 1 μM, as expected, sensors (1) and (2) showed weak fluorescence in aqueous solution. Upon addition of different metal ions, sensors (1) and (2) showed remarkable fluorescence enhancement only with Cu2+, due to coordination to a paramagnetic Cu2+ centre (Fig. 1A and 2A).
:1, v/v) solution [sensor (1) and (2)] = 1 and 2 μM, [Cu2+] = 1 μM, as expected, sensors (1) and (2) showed weak fluorescence in aqueous solution. Upon addition of different metal ions, sensors (1) and (2) showed remarkable fluorescence enhancement only with Cu2+, due to coordination to a paramagnetic Cu2+ centre (Fig. 1A and 2A).
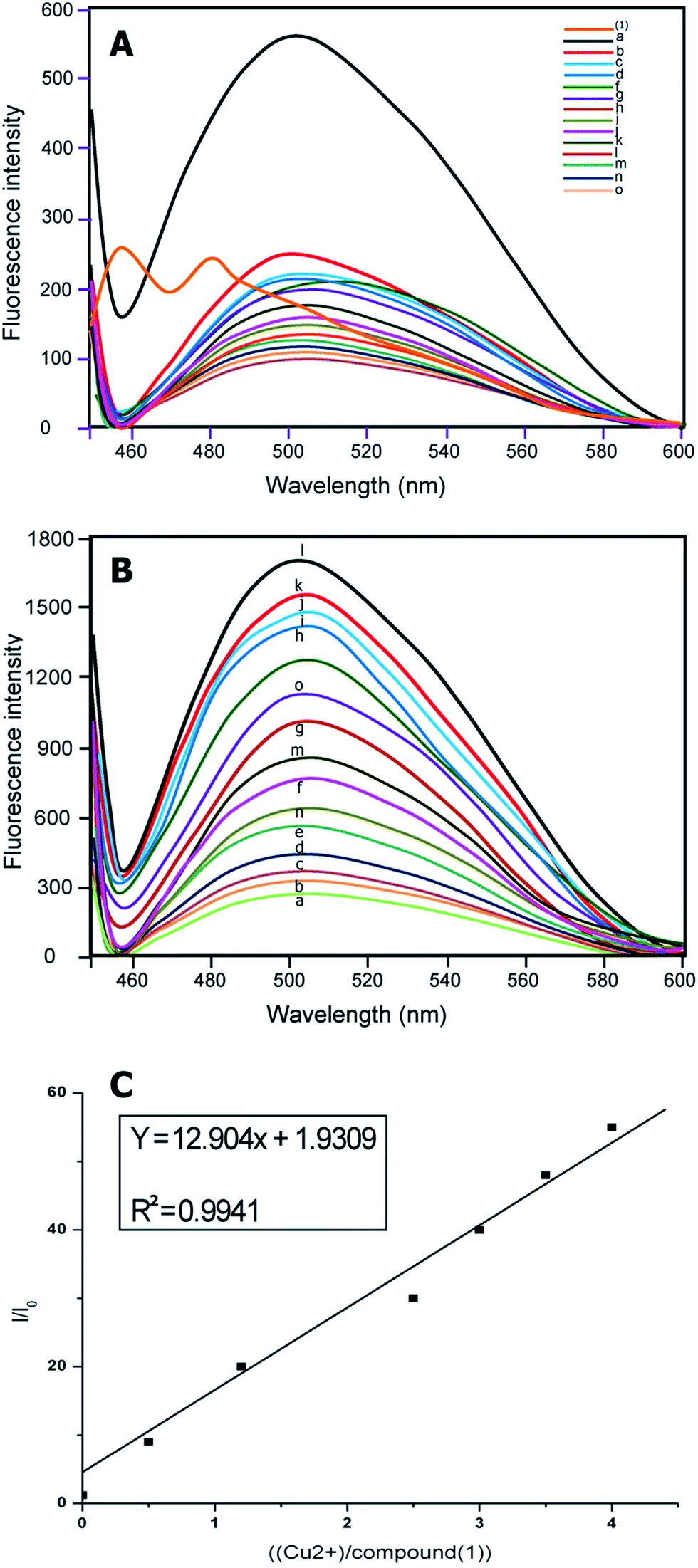 | ||
| Fig. 1 (A) Fluorescence emission spectra of chemosensors for compound (1) (1 μmol L−1) with various metal ions (perchlorate, chloride, or nitrate salts of (a) Cu2+, (b) Ni2+, (c) Co2+, (d) Zn2+, (e) Pb2+, (f) Hg2+, (g) Mg2+, (h) Cr3+, (i) Na+, (j) K+, (k) Fe2+, (l) Fe3+, (m) Al3+, (n) Cd2+ and (o) Ag+) in aqueous CH3CH2OH (CH3CH2OH–H2O = 1:1, v/v, 10 μM HEPES buffer, pH = 7.0). (B) Emission spectra of compound (1) in the presence of an increasing concentration of Cu2+/(μmol L−1) from a to o, that is, 0, 0.2, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, 1.8, 2.0, 2.5, 3.5, 4.0, 4.5 respectively, 1 μmol L−1 compound (1) in 1:1 (volume ratio) CH3CH2OH–H2O (10 mmol L−1 10 μM HEPES buffer, pH = 7.0) binding to Cu2+ with different concentrations. (C) Fluorescence enhancement relationship of I/I0 with [Cu2+]/[compound (1)]. | ||
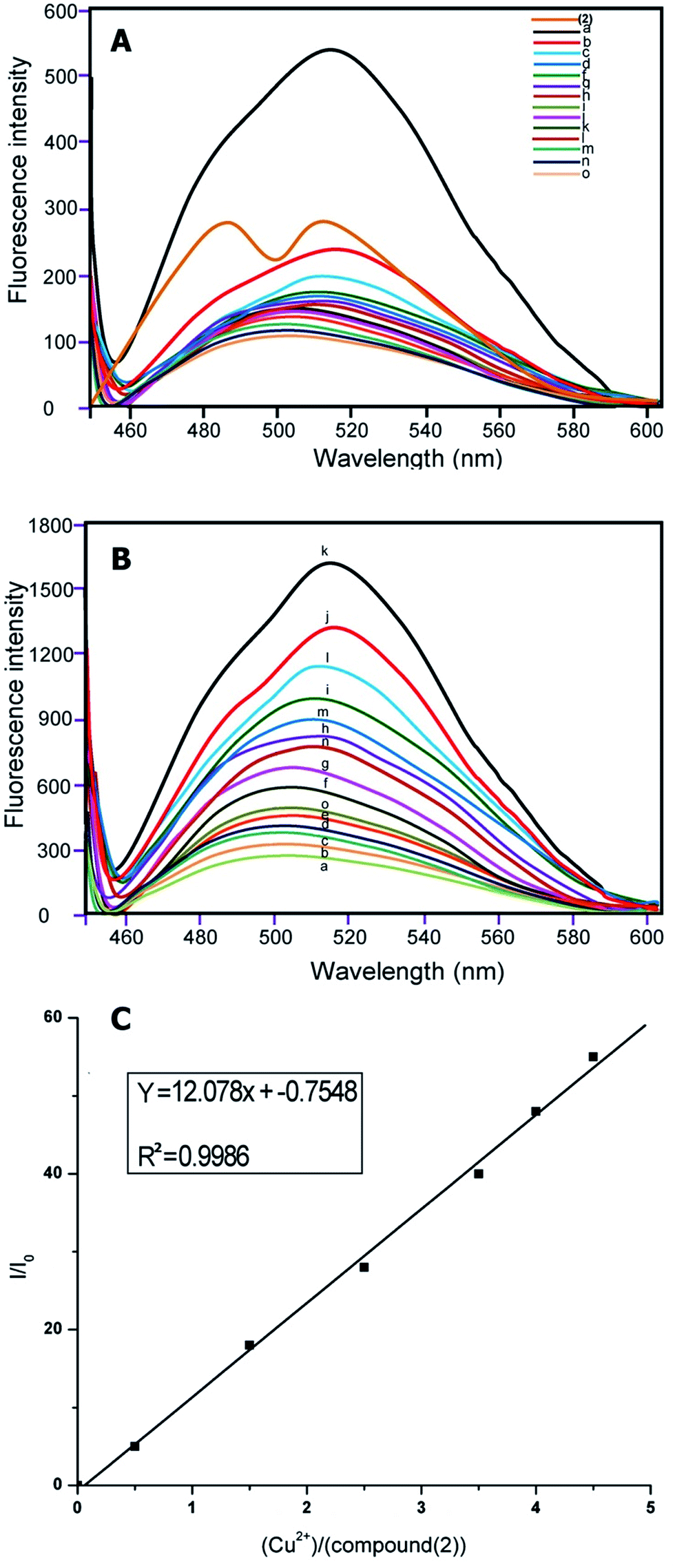 | ||
| Fig. 2 (A) Fluorescence emission spectra of 10 μm solution chemosensors for compound (2) with various metal ions perchlorate, chloride, or nitrate salts of (a) Cu2+, (b) Ni2+, (c) Co2+, (d) Zn2+, (e) Pb2+, (f) Hg2+, (g) Mg2+, (h) Cr3+, (i) Na+, (j) K+, (k) Fe2+, (l) Fe3+, (m) Al3+, (n) Cd2+, and (o) Ag+ in aqueous CH3CH2OH (CH3CH2OH–H2O = 4/1, v/v, 10 μM HEPES buffer, pH = 7.0). (B) Emission spectra of compound (2) in presence of increasing concentration of Cu2+/(μmol L−1) from a to o, that is, 0, 0.2, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, 1.8, 2.0, 2.5, 3.5, 4.0, 4.5 respectively, 1 μmol L−1 compound (2) in 1:1 (volume ratio) CH3CH2OH–H2O (10 mmol L−1 10 μM HEPES buffer, pH = 7.0) binding to Cu2+ with various ion concentrations. (C) Fluorescence enhancement relationship of I/I0 with [Cu2+]/[compound (2)]. | ||
Based on earlier studies37–39 and our own observations, we proposed the following mechanism. Cu2+ is a well-known paramagnetic ion with an unfilled d shell and could strongly quench the fluorescence of the fluorophore near it via electron or energy transfer. Thus it was of interest that the Cu2+ by compounds (1) and (2) did not quench the fluorescence. It is known that the nitrogen lone pair electrons in compounds (1) and (2) can quench the fluorescence of the indene 1,3-dione moiety through photo-induced electron transfer (PET).40–44 Masking of the nitrogen lone-pair electrons caused suppression of their fluorescence quenching, and therefore, results increased in fluorescence intensity. It is evident from the emission spectra that in the absence of Cu2+ the sensor exhibited weak fluorescence owing to quenching of compounds (1) and (2) fluorescence through PET. Also, in the presence of the quenching metal ions, the relative contribution of two opposing factors, the metal-ion receptor binding that led to enhancement of the fluorescence signal and the fluorophore–metal ion interaction that led to fluorophore quenching determined the net effect. Therefore, it was possible to observe fluorescence enhancement in the presence of quenching ions when metal binding-induced enhancement of fluorescence intensity was greater than the quenching-induced reduction in fluorescence intensity. So, it is clear that for an already PET-quenched system, otherwise strong-quenching metal ions act as poor quenchers. Obviously, in the absence of quenching interaction, metal ions binding with receptors will only lead to fluorescence enhancement. Finally, we conclude that unavailability of the lone-pair electrons of N for PET caused fluorescence enhancement.
The emission at around 500 nm in (1) and 510 nm in (2) did not change when the concentration of Cu2+ increased from 1 μmol L−1 of compounds (1) and (2) in 1:1 (volume ratio) CH3CH2OH–H2O (10 mmol L−1 10 μM HEPES buffer, pH = 7.0) binding to Cu2+ with different ion concentrations.
Implied here is a 1:1 complex of compounds (1) and (2) with Cu2+ ions (Fig. 1B and 2B). The maximum fluorescence enhancement was observed in the presence of 2 μm (1) and 1.8 μm in (2). When the concentration of Cu2+ was less or equal to around 2 μm, the binding of Cu2+ made the polarization of the molecule larger, and it allowed only partial electron transfer; therefore the fluorescence intensity was enhanced but the emission maximum did not change with the increase of Cu2+ concentration. When the concentration of Cu2+ was more than 2 μm, excess Cu2+ resulted in a sudden drop of relative intensity, no doubt due to concentration quenching.45
Furthermore, the relationship of fluorescence enhancement ratio (defined as I/I0, where I and I0 are the fluorescence intensities of compound (1) and (2) in solution when binding with and without Cu2+, respectively) with the binding concentration of Cu2+ was also investigated. Apparently, the ratio was nearly proportional to the molar binding concentration of Cu2+ (R2 = 0.9941 (1) and R2 = 0.9986 (2)) when the binding concentration of copper was in a range of 0–5.0 μmol L−1 (Fig. 1c and 2c).
pH-dependent behavior
The influence of pH on chemosensor of compounds (1) and (2) were studied using UV-vis-spectrocopy (Fig. S11†). Over a pH range of 3–8, the visible absorption bands centered at 480 nm in (1) and 450 nm in (2) were unchanged. But increasing pH from 9 to 11 engendered a shift in the maximum absorption wavelength to 375 nm (1) and 380 nm (2) with color change of yellow to blue. This difference was due to dissociation of the (1)–Cu2+ and (2)–Cu2+ complex, which resulted in lower absorbance and color change from yellow to blue.The pH emission spectra of sensors (1) and (2) and its ability to detect the Cu2+ cations might be influenced by the solution pH because of the nitrogen atom in hydrazone moiety with lone-pair electrons in the sensors (1) and (2). To verify this hypothesis, the pH effect on the fluorescence intensity of sensors (1) and (2) in unbound and bound Cu2+ was studied. As shown in Fig. S12,† in the unbound Cu2+ (curve b), sensors (1) and (2) presented a low fluorescence intensity that remained stable and did not change with the pH. Upon addition of Cu2+ (curve a), the emission intensity of compounds (1) and (2) with Cu2+ increased dramatically from pH 3 to 5, resulting from competition between the N–H proton and Cu2+ ion.46,47 In particular, slight significant changes in fluorescence spectra were observed in the pH 5–8 range and decreased under alkaline conditions with a color change of yellow to blue. The quenching at higher pH could be well explained by formation of Cu(OH)− or Cu(OH)2, thus reducing the concentration of Cu2+–(1)/(2);48 therefore, there were fewer ions as Cu2+ in solution that were able to be complexed with compounds (1) and (2), which explains the decrease in fluorescence intensity. The results demonstrated that sensors (1) and (2) could be used in environments where pH ranges from 5 to 8, which cover physiological pH conditions.
Elemental mapping FE-SEM was used to confirm the presence of C, O, N, and Cu in the surface of complexes (corresponding to compounds (1) and (2)). Fig. 3a and 4a exhibit FE-SEM color images of complexes (compounds (1)–Cu2+ and (2)–Cu2+); Fig. 3b–e and 4b–e show the elemental mapping for carbon, nitrogen, oxygen, and copper, respectively. It is evident from Fig. 3b–d and 4b–d that carbon, nitrogen, and oxygen are higher in density. There is a homogenous distribution of carbon, nitrogen, oxygen, and copper (Fig. 3a and 4a). Thus, elemental mapping shows that the complex of compounds (1) and (2) contains Cu2+. This also indicates the purity of the complexes (compounds (1)–Cu2+ and (2)–Cu2+). EDS is generally accurate up to trace amounts of metal present in the surface of complexes. The EDS recorded from the selected area is shown in Fig. 3f and 4f, which reveals the presence of C, O, N, and Cu in the complexes.
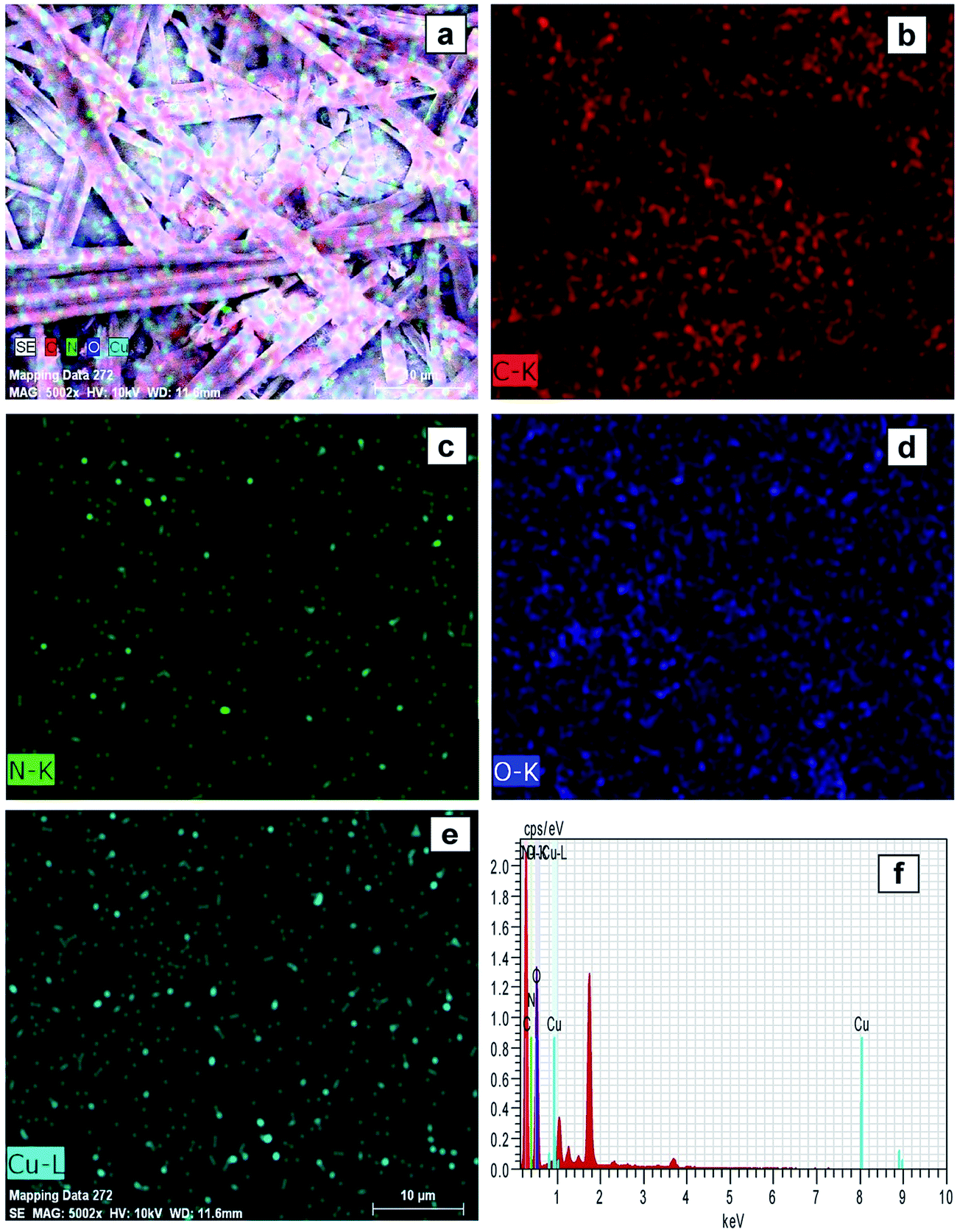 | ||
| Fig. 3 FE-SEM elemental colour mapping image of compound (1)–Cu2+. (a) Survey spectrum, (b) C, (c) N, (d) O, (e) Cu, and (f) EDS spectra. | ||
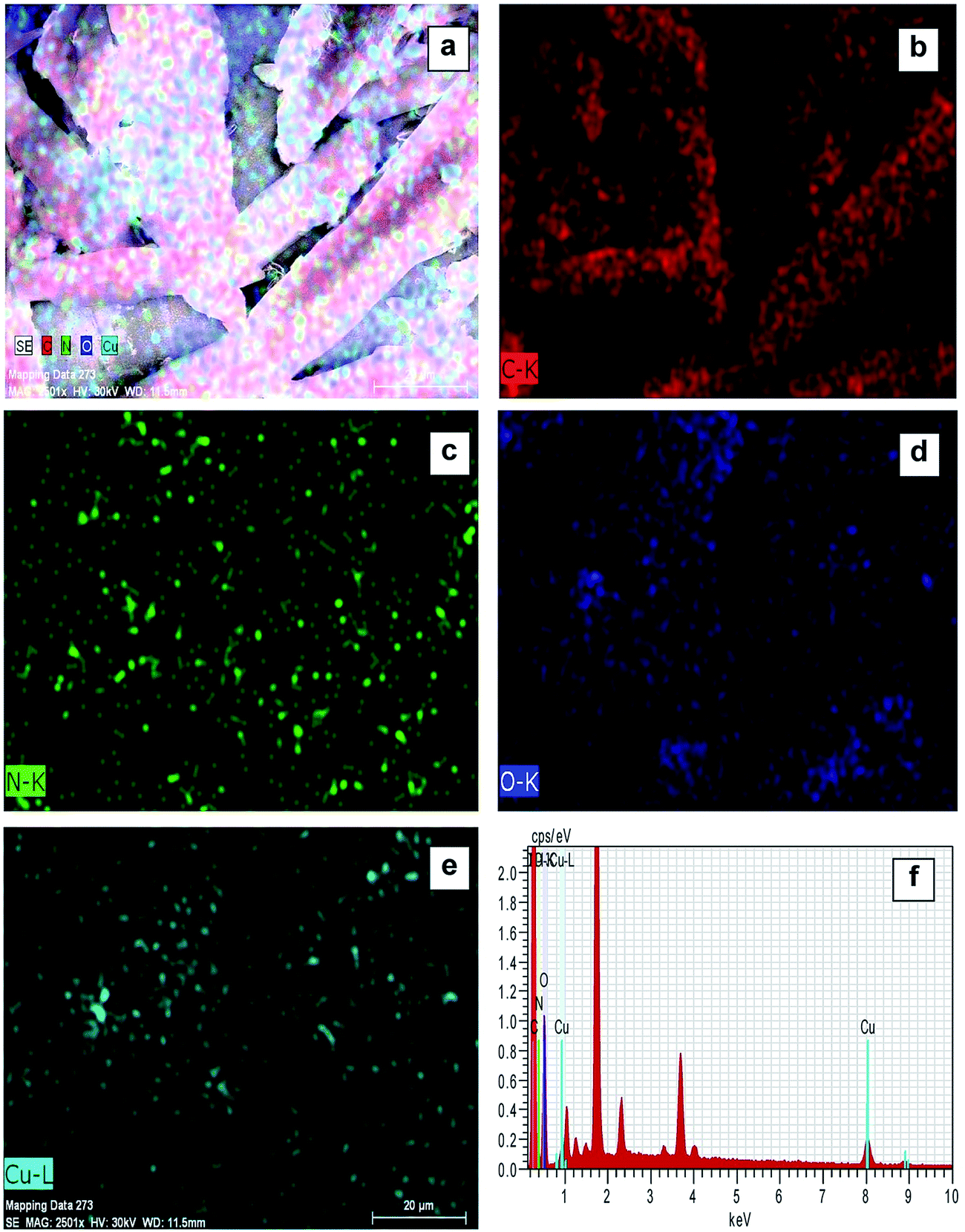 | ||
| Fig. 4 FE-SEM elemental colour mapping image of compound (2)-Cu2+. (a) Survey spectrum, (b) C, (c) N, (d) O, (e) Cu, and (f) EDS spectra. | ||
Docking studies
We studied the 4LRH, 4L9K, 4EKD, and 4GIW cancer proteins from the protein data bank. Among these proteins, only 4LRH showed good results compared to other binding energy values. Molecular docking simulations were employed to predict interaction of compounds (1) and (2) with 4LRH. We docked compounds (1) and (2) with 4LRH as shown in Fig. 5 and 6. The value of free-energy binding (ΔE) and the binding constant (ki) of compounds (1) and (2) are given in Table 1. The molecular docking simulation shows that compound (1) has less activity compared with compound (2). The above conclusion is proved from hydrogen bond formations. The simulations for compounds (1) and (2) were carried out with three other cancer proteins (4L9K, 4EKD, and 4GIW) as shown in Fig. S17–S22.† As seen in the figures, it is evident that 4LRH docks strongly with compounds (1) and (2). The hydrogen bond distance between compound (2) and residues was smaller than the hydrogen bond distance between compound (1) and residues. In addition, hydrophobic interaction of compound (2)-4LRH was stronger than compound (1)-4LRH. The docked pose displayed in Fig. 5a and b and 6a and b reveals the protein subdomain IB to be the favorable binding site for the drug. The principal hydrophobic binding sites in 4LRH are located in domains II and III, while domain I, characterized by a net negative charge, normally serves as a suitable binding site for cationic probe molecules. The lowest binding energy conformer was obtained from four different conformers for every docking simulation and the consequential minimum energy conformation was used for further analysis. As is usual in a blind docking simulation protocol, we obtained a number of binding sites and the corresponding binding constants and free energies. Compelling evidence for the probable-binding location of the drug in subdomain IB of 4LRH was derived from the observation that the binding of compounds (1) and (2) in the IB region was found to be characterized by a favorable binding energy of −8.25 kcal mol−1 and −8.29 kcal mol−1, respectively (Table 1), a very low inhibition constant (900.66 nM) for compound (1) and (837.70 nM) for compound (2).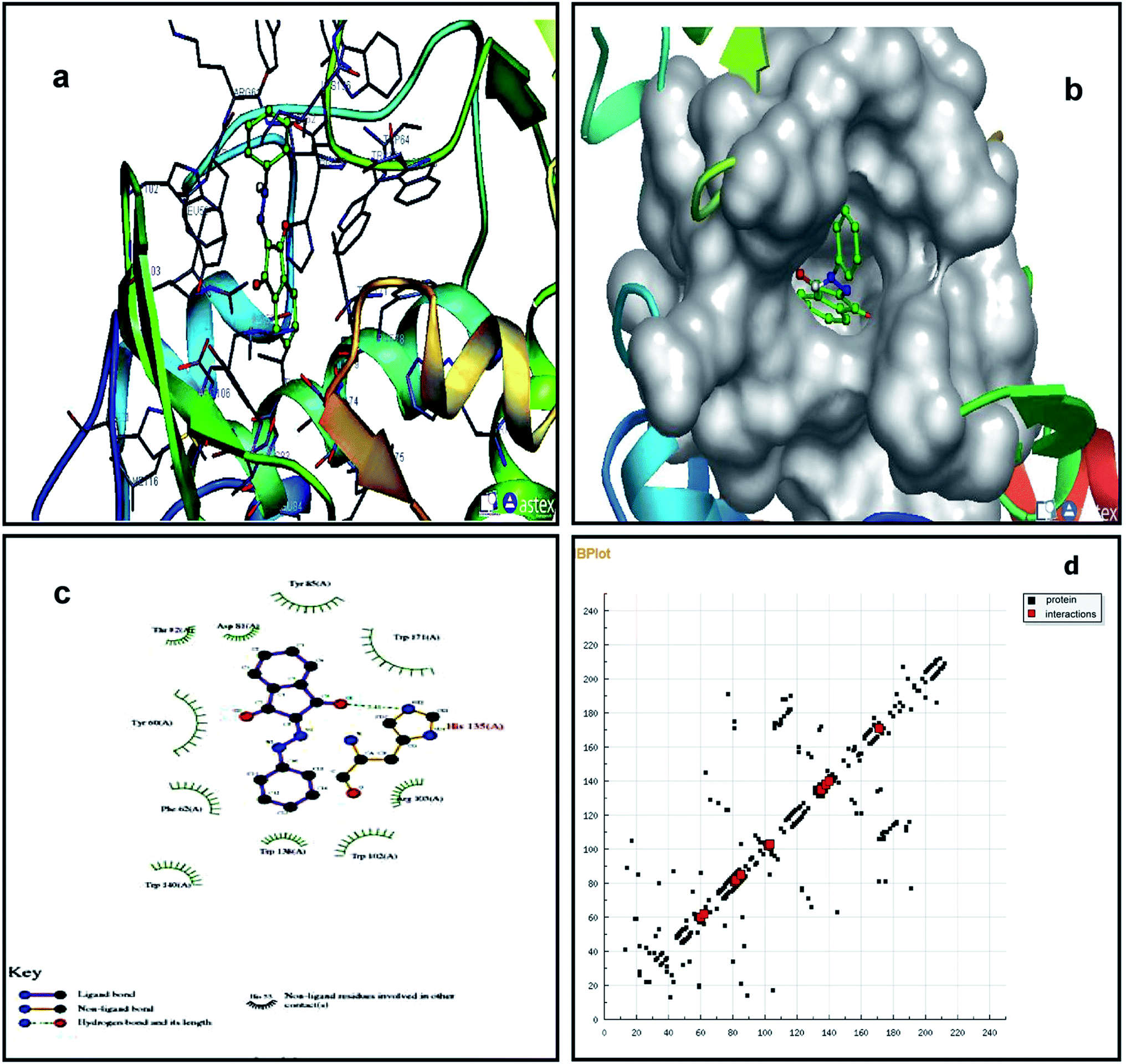 | ||
| Fig. 5 Molecular docking studies of compound (1) with 4LRH. | ||
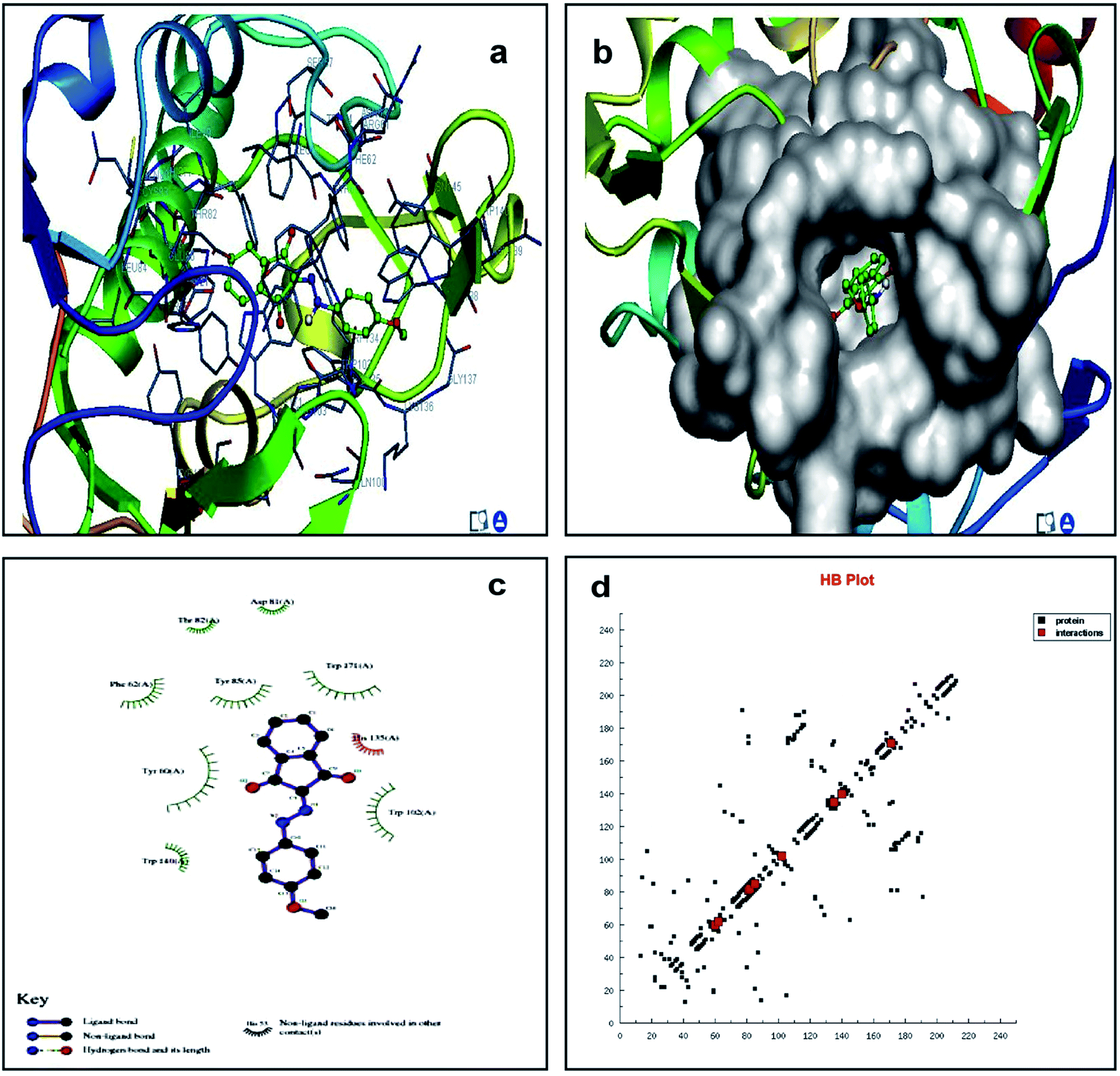 | ||
| Fig. 6 Molecular docking studies of compound (2) with 4LRH. | ||
Fig. 5c shows the protein residues in the near vicinity (within 4 Å) of the probe, which shows the presence of both hydrophobic residues (TYR-60, TRP-171, and TYR-85), as well as charged/polar residues (TRP-102 (cation-pi), TRP-140, and HIS-135) for compound (1) and hydrophobic residues (TRP-171, TYR-85, PHE-62, TRP-140, and TRP-138), as well as charged/polar residues (TYR-60 (cation-pi), HIS-135 (hydrogen bond), ARG-103) for compound (2) (Fig. 6c). These results indicate that the binding phenomenon was mainly governed by hydrophobic forces with a significant contribution from electrostatic interactions. Asymmetric charge distribution over the molecule due to the presence of heteroatoms might have been responsible for such binding. Based on molecular docking results, we conclude that the alkyl group has been more responsible for biological activity than the aromatic ring.
Cytotoxicity
The cytotoxicity responses of compounds (1) and (2) in various concentrations added to cancer cells are clearly evident in cellular imaging. Hence, results indicated that compounds (1) and (2) are efficient candidates for monitoring changes in intracellular concentrations under certain biological conditions. To test cytotoxicity, we performed MTT assays in KB cancer cells treated with various concentrations of compounds (1) and (2) for up to 5 h. As shown in Fig. 7 and 8, compounds (1) and (2) showed significant cytotoxic effects on KB cancer cells for at least up to 4 h. The synthesized compounds (1) and (2) were examined for their cytotoxic properties on the KB cell line by means of MTT test, which allowed us to evaluate the toxic effect of the compounds on cellular mitochondrial metabolism. Cells were tested for 48 h with increasing concentrations of tested compounds. Microscopic images of control cancer cells and apoptotic morphological changes in the KB cell line treated with compounds (1) and (2) are shown in Fig. 7 and 8, respectively. The results indicate that compound (1) showed minimal cell death compared to compound (2). Compounds (1) and (2) exhibited broad inhibition on KB cell lines with IC50 values of 32.78 and 40.08, respectively. The IC50 values of the compound (Fig. S23†) suggest that compound (2) had a greater inhibitory effect on the cancer cells. Compound (2) carried the –OCH3 group in para position and showed the highest IC50 value, convincing us to propose that the electronic effect may be one of the factors in determining the anticancer action of compound (2).49,50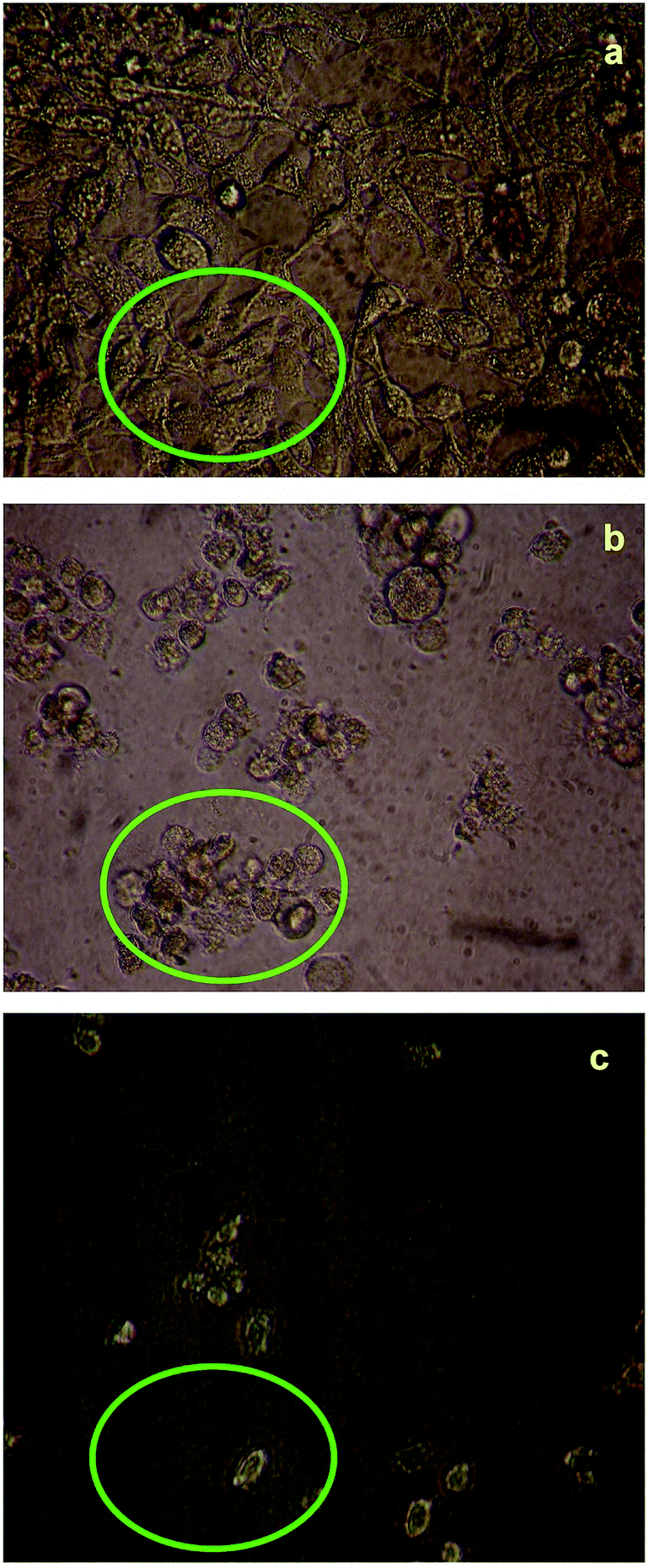 | ||
| Fig. 7 Live cell images of compound (1): (a) before and (b and c) after treatment with compound (1), examined by fluorescence microscopy. | ||
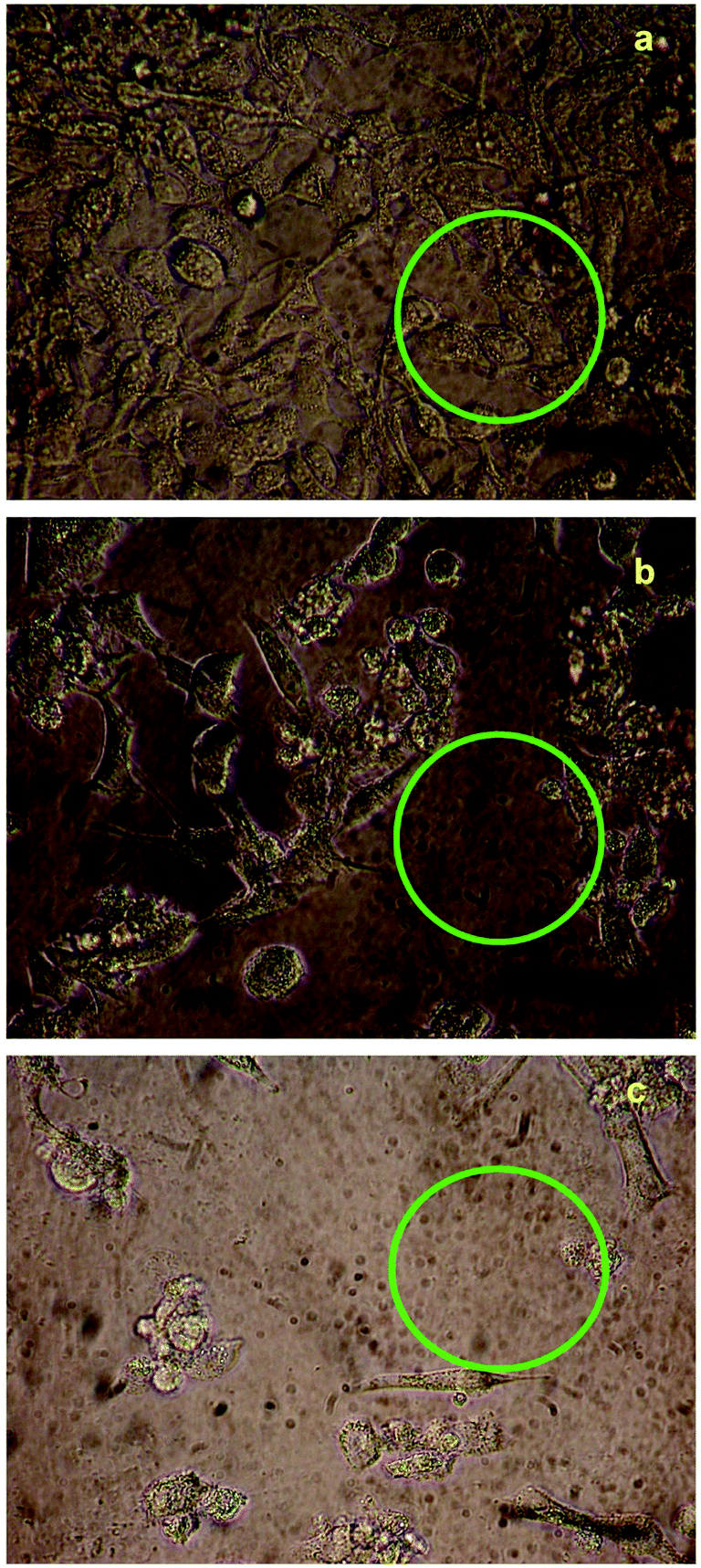 | ||
| Fig. 8 Live cell images of compound (2): (a) before and (b and c) after treatment with compound (1), examined by fluorescence microscopy. | ||
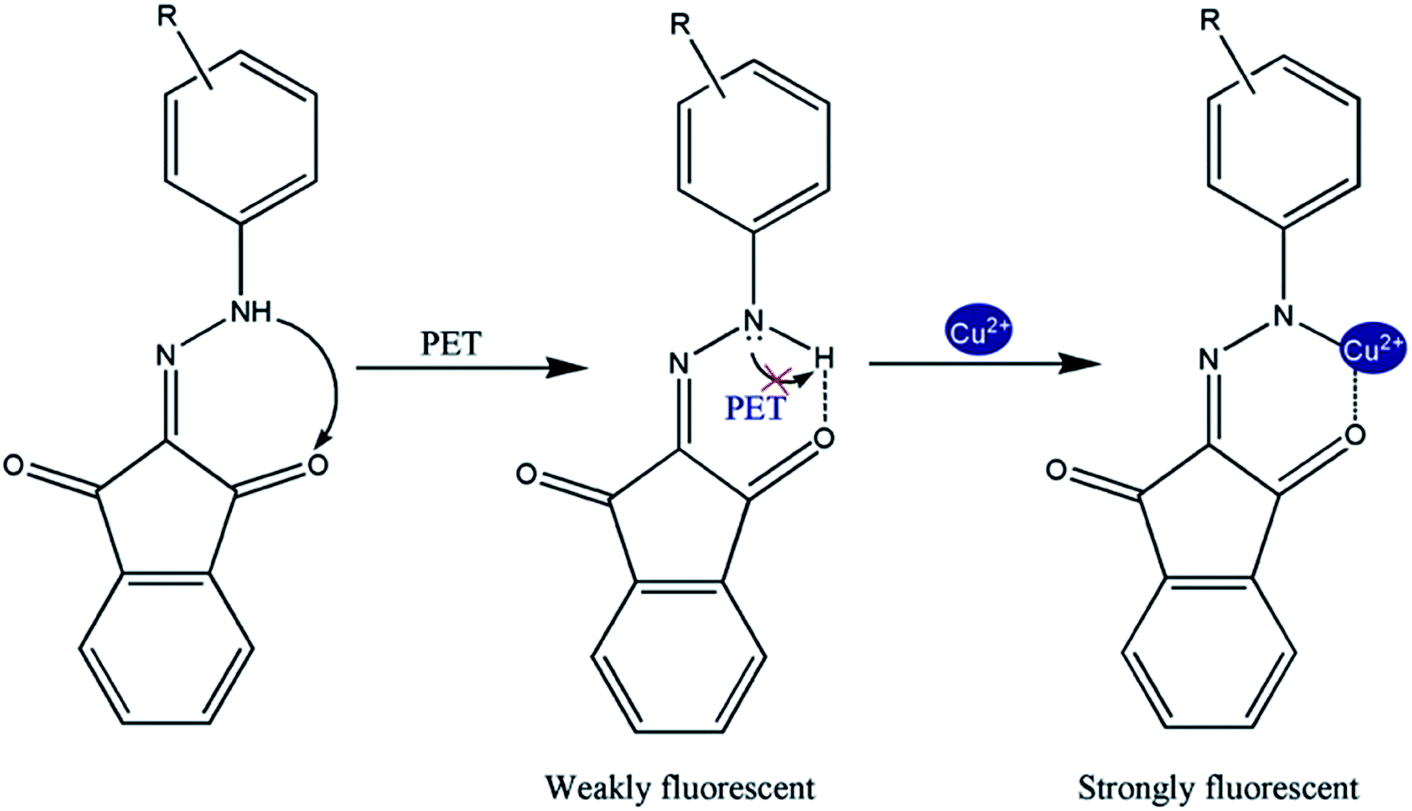 | ||
| Fig. 9 Photoinduced electron transfer (PET) of compounds (1) and (2) with Cu2+. | ||
Theoretical calculations
The geometries of sensor compounds (1) and compound (2) with the Cu2+ ion were optimized using DFT-B3LYP 6-31G and LANL2DZ (d) levels, respectively, using the Gaussian 03 package. In Fig. S24 and S25,† the optimized geometries of compound (1), compound (2), and the 1:1 complex with Cu2+ ion are given, which display the effective binding sites, namely –NH and CO for the Cu2+ ion. To obtain insight into electronic behavior in the presence and absence of Cu2+ with compound (1) and compound (2), TD-DFT calculations were carried out. HOMO and LUMO plots for compound (2) showed that the π-electrons on the ligand HOMO focused on one of the ketone groups of the dione moiety and the ligand LUMO focused on the N–NH group of the hydrazone moiety. When Cu2+ was added, it showed a significant change in the distribution of the π-electrons on the HOMO and LUMO ligands. More significantly, the HOMO and LUMO densities in compounds (1) and (2) with Cu2+ were delocalized over the entire molecule except for the phenyl ring of the dione moiety in HOMO and the methoxy phenyl ring in LUMO. The energies of HOMO and LUMO of compounds (1) and (2)–Cu2+ complex were slightly lower than that of compounds (1) and (2). More importantly, the decreasing energy in the LUMO was more obvious than that of the HOMO. It suggests a smaller HOMO–LUMO gap and a bathochromic shift compared with compounds in emission, which may result from the increased conjugation of the complex.
Conclusions
In summary, the hydrazones of indene-1,3-dione have been synthesized and characterized in IR, 1H, 13C NMR spectra studies. The FT-IR, NMR comprised a promising analytical approach for detecting Cu2+ in ethanol–water solutions. When copper ions were added in (1) and (2), morphology changes were observed. We prepared a simple type of fluorescent “turn-on” chemosensor based on hydrazones that shows interesting properties such as high sensitivity to Cu2+. It possesses a high affinity and selectivity for copper ions relative to most other competitive metal ions via enhancement of the monomer fluorescence emission of hydrazones in organic aqueous solutions. We expect that the present design strategy and the remarkable photophysical properties of this sensor will help to extend applications of fluorescent sensors for metal ions. Regarding compounds (1) and (2) docking with 4LRH, 4L9K, 4EKD and 4GIW proteins, only 4LRH cancer protein showed good results when compared to other proteins' binding energy values. Compounds (1) and (2) have potent in vitro cytotoxicity against the KB cell line. In the present investigation, molecular structure, HOMO, LUMO, Mulliken charges and MEP analysis were examined using ab initio DFT B3LYP/6-31G calculations.Notes and references
- K. Dhara, U. C. Saha, A. Dan, M. Manassero, S. Sarkar and P. Chattopadhyay, Chem. Commun., 2010, 46, 1754 RSC.
- U. C. Saha, B. Chattopadhyay, K. Dhara, S. K. Mandal, S. Sarkar, A. R. Khuda-Bukhsh, M. Mukherjee, M. Helliwell and P. Chattopadhyay, Inorg. Chem., 2011, 50, 1213 CrossRef CAS PubMed.
- H. H. Harris, I. Pickering and G. N. George, Science, 2003, 301, 1203 CrossRef CAS PubMed.
- P. B. Tchounwou, W. K. Ayensu, N. Ninashvile and D. Sutton, Environ. Toxicol., 2003, 18, 149 CrossRef CAS PubMed.
- Y. Ronghua, L. Kean, W. Kemin, Z. Fenglin, L. Na and L. Feng, Anal. Chem., 2003, 75, 612 CrossRef.
- L. B. Desmonts, D. N. Reinhoudt and M. C. Calama, Chem. Soc. Rev., 2007, 36, 993 RSC.
- H. H. Wang, L. Xue, Z. J. Fang, G. P. Li and H. Jiang, New J. Chem., 2010, 34, 1239 RSC.
- (a) R. Kramer, Angew. Chem., Int. Ed., 1998, 37, 772 CrossRef CAS; (b) K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205 CrossRef CAS PubMed; (c) E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517 CrossRef CAS PubMed; (d) G. L. Millhauser, Acc. Chem. Res., 2004, 37, 79 CrossRef CAS PubMed; (e) E. Gaggelli, H. Kozlowski and G. Valensin, Chem. Rev., 2006, 106, 1995 CrossRef CAS PubMed; (f) G. Multhaup, A. Schlicksupp, L. Hess, D. Beher, T. Ruppert, C. L. Masters and K. Beyreuther, Science, 1996, 271, 1406 CAS; (g) R. A. Lovstad, BioMetals, 2004, 17, 111 CrossRef CAS.
- G. F. Nordberg, R. F. M. Herber and L. Alessio, Cadmium in the Human Environment, Oxford University Press, Oxford, UK, 1992 Search PubMed.
- C. N. McFarland, L. I. Bendell-Young, C. Guglielmo and T. D. Williams, J. Environ. Monit., 2002, 4, 791 RSC.
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210 RSC.
- Z. Li, W. Zhao, X. Li, Y. Zhu, C. Liu, L. Wang, M. Yu, L. Wei, M. Tang and H. Zhang, Inorg. Chem., 2012, 51, 12444 CrossRef CAS PubMed.
- J. W. Liu and Y. Lu, J. Am. Chem. Soc., 2007, 129, 9838 CrossRef CAS PubMed.
- K. M. K. Swamy, S. K. Ko, S. K. Kwon, H. N. Lee, C. Mao, J. M. Kim, K. H. Lee, J. Kim, I. Shin and J. Yoon, Chem. Commun., 2008, 5915 RSC.
- A. Sigel, H. Sigel and R. K. O. Sigel, Nickel and Its Surprising Impact in Nature; Metal Ions in Life Sciences, Wiley, Chichester, U.K., 2007, vol. 2 Search PubMed.
- (a) A. Banerjee, A. Sahana, S. Guha, S. Lohar, I. Hauli, S. K. Mukhopadhyay, J. Matalobos and D. Das, Inorg. Chem., 2012, 51, 5699 CrossRef CAS PubMed; (b) G. Li, H. Fang, Y. Cai, Z. Zhou, P. K. Thallapally and J. Tian, Inorg. Chem., 2010, 49, 7241 CrossRef CAS PubMed; (c) S. C. Dodani, Q. He and C. J. Chang, J. Am. Chem. Soc., 2009, 131, 18020 CrossRef CAS PubMed.
- (a) A. N. Anthemidis and C. P. Karapatouchas, Microchim. Acta, 2008, 160, 455 CrossRef CAS; (b) G. Kaya and M. Yaman, Talanta, 2008, 75, 1127 CrossRef CAS PubMed.
- A. C. Davis, C. P. Calloway Jr and B. T. Jones, Talanta, 2007, 71, 1144 CrossRef CAS PubMed.
- (a) X. C. Wang, S. H. Drew, K. J. Lee, J. K. Senecal and L. A. Samuelson, Nano Lett., 2002, 2, 1273 CrossRef CAS; (b) R. Metivier, I. Leray and B. Valeur, Chem.–Eur. J., 2004, 10, 4480 CrossRef CAS PubMed.
- H. Tian, B. Li, J. Zhu, H. Wang, Y. Li, J. Xu, J. Wang, W. Wang, Z. Sun, W. Liu, X. Huang, X. Yan, Q. Wang, X. Yao and Y. Tang, Dalton Trans., 2012, 41, 2060 RSC.
- Y. Zhao, X. B. Zhang, Z. X. Han, L. Qiao, C. Y. Li, L. X. Jian, G. L. Shen and R. Q. Yu, Anal. Chem., 2009, 81, 7022 CrossRef CAS PubMed.
- H. H. Wang, L. Xue, Z. J. Fang, G. P. Li and H. Jiang, New J. Chem., 2010, 34, 1239 RSC.
- F. J. Huo, C. X. Yin, Y. T. Yang, J. Su, J. B. Chao and D. S. Liu, Anal. Chem., 2012, 84, 2219 CrossRef CAS PubMed.
- V. Chandrasekhar, S. Das, R. Yadav, S. Hossain, R. Parihar, G. Subramaniam and P. Sen, Inorg. Chem., 2012, 51, 8664 CrossRef CAS PubMed.
- Z. Li, W. Zhao, X. Li, Y. Zhu, C. Liu, L. Wang, M. Yu, L. Wei, M. Tang and H. Zhang, Inorg. Chem., 2012, 51, 12444 CrossRef CAS PubMed.
- T. Mistri, R. Alam, M. Dolai, S. K. Mandal, A. R. K. Bukhsh and M. Ali, Org. Biomol. Chem., 2013, 11, 1563 CAS.
- J. T. Yeh, W. C. Chen, S. R. Liu and S. P. Wu, New J. Chem., 2014, 38, 4434 RSC.
- S. Anbu, R. Ravishankaran, M. F. C. Guedes da Silva, A. A. A. Karande and A. J. L. Pombeiro, Inorg. Chem., 2014, 53, 6655 CrossRef CAS PubMed.
- F. R. Pavan, P. I. S. Maia, S. R. A. Leite, V. M. Deflon, A. A. Batista, D. N. Sato, S. G. Franzblau and C. Q. F. Leite, Eur. J. Med. Chem., 2010, 45, 1898 CrossRef CAS PubMed.
- G. Gilli and P. Gilli, The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory, Oxford University Press, Oxford, 2009 Search PubMed.
- J. R. Heldt, J. Heldt, M. Jozefowicz and J. Kaminski, J. Fluoresc., 2001, 11, 65 CrossRef CAS.
- L. A. Estrada, J. E. Yarnell and D. C. Neckers, J. Phys. Chem. A, 2011, 115, 6366 CrossRef CAS PubMed.
- P. Ashokekumar, V. Thiagarajan, S. Vasanthi and P. Ramamurthy, J. Photochem. Photobiol., A, 2009, 208, 117 CrossRef PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 2nd edn, 1999 Search PubMed.
- M. W. Wong, M. J. Frisch and K. B. Wiberg, J. Am. Chem. Soc., 1991, 113, 4776 CrossRef CAS.
- P. Gilli and G. Gilli, J. Mol. Struct., 2010, 972, 2 CrossRef CAS PubMed.
- A. P. de silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed.
- A. P. de silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385 RSC.
- A. P. de silva, A. Goligher, H. Q. N. Gunaratne and T. E. Rice, Arkivoc, 2003, 8, 229 CrossRef.
- Z. Xu, J. Yoon and D. R. Spring, Chem. Commun., 2010, 46, 2563 RSC.
- Z. Xu, Y. Xiao, X. Qian, J. cui and D. Cui, Org. Lett., 2005, 7, 889 CrossRef CAS PubMed.
- W. Wang, G. Sprigsteen, S. Gao and B. Wang, Chem. Commun., 2000, 1283 RSC.
- B. Ramachandram and A. Samanta, Chem. Commun., 1997, 1037 RSC.
- B. Ramachandram and B. Samanta, J. Phys. Chem. A, 1998, 102, 10579 CrossRef CAS.
- K. T. Mahmudov, A. M. Maharramov, R. A. Aliyeva, F. M. Chyragov, R. K. Askerov, P. Q. Hasanov, M. N. Kopylovich and A. J. L. Pombeiro, J. Mol. Struct., 2011, 1006, 576 CrossRef CAS PubMed.
- L. Xue, G. P. Li, D. J. Zhu, Q. Liu and H. Jiang, Inorg. Chem., 2012, 51, 10842 CrossRef CAS PubMed.
- Y. Zhang, X. F. Guo, W. X. Si, L. H. Jia and X. H. Qian, Org. Lett., 2008, 10, 473 CrossRef CAS PubMed.
- X. Y. Zhou, B. R. Yu, Y. L. Guo, X. L. Tang, H. H. Zhang and W. S. Liu, Inorg. Chem., 2010, 49, 4002 CrossRef CAS PubMed.
- (a) Z. Dai and J. W. Canary, New J. Chem., 2007, 31, 1708–1718 RSC; (b) B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3 CrossRef CAS.
- M. Guerrero, J. Pons, J. Ros, M. F. Bardia, O. Vallcorba, J. Rius, V. Branchadell and A. Merkoci, CrystEngComm, 2011, 13, 6457 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11006d |
| This journal is © The Royal Society of Chemistry 2014 |