
Cross-metathesis approach to produce precursors of nylon 12 and nylon 13 from microalgae†
Godwin Ameh Abel‡
a,
Kim Oliver Nguyen‡§
b,
Sridhar Viamajalaac,
Sasidhar Varanasiac and
Kana Yamamoto*bc
aDepartment of Chemical and Environmental Engineering, University of Toledo, 1640 Westwood Ave, Toledo, OH 43606, USA
bDepartment of Chemistry and Biochemistry, University of Toledo, 2801 W. Bancroft St, Toledo, OH 43606, USA. E-mail: kana.yamamoto@utoledo.edu
cSchool of Green Chemistry and Engineering, University of Toledo, Toledo, OH 43606, USA
First published on 6th October 2014
Abstract
A two-step synthesis for producing methyl 12-aminododecanoate and 13-aminotridecanoate, the precursors of nylon 12 and nylon 13, from methyl oleate is described. First, methyl 11-cyano-9-undecenoate or 12-cyano-9-dodecenoate were prepared by cross metathesis of methyl oleate with either allyl cyanide or homoallyl cyanide, respectively. Subsequently, all the unsaturation of the two intermediates was hydrogenated to deliver the final products. This method represents the first synthesis of nylon 12 and 13 precursors from methyl oleate, an ester of an abundant and renewable natural fatty acid present in vegetable oil or microalgae. It also represents the shortest synthesis of nylon precursors from fatty acids, and as demonstrated in this study, can be directly applied to crude fatty acid methyl ester extracts from microalgae.
Introduction
Nylons are synthetic polyamides with extensive applications in modern society. In particular, nylon 11 and 12 possess excellent chemical properties and are widely used in the automotive, medical, and electronic industries as well as for sports equipment, back panels for solar cells, and lenses for glasses. Nylon 13 is reported to have similar properties to nylon 12. These nylons are produced from amino acids or their derivatives with a corresponding number (11, 12 or 13) of carbon-chain backbone.1Currently, the precursor of nylon 12 is manufactured from petroleum-derived butadiene in a six-step process.2 However, there is an increasing interest in use of renewable sources for production of these amino acid precursors, because of concerns over environmental sustainability of petrochemical products.3 As such, synthetic approaches that use natural fatty acids and esters from plant- or algae-derived feedstocks are especially attractive.4–6 Among the natural fatty acids, oleic acid is the predominant component of lipids in a large variety of vegetable oils (e.g. soy or canola) as well as oleaginous microorganisms such as algae.7
The precursor of nylon 11 is manufactured from ricinoleic acid, a natural fatty acid available only in castor beans.2 In this method, the acid is first converted to its methyl ester and then subjected to pyrolysis to produce 10-undecyleate, which is then converted to the nylon 11 precursor in two additional steps. Several other multi-step (>4 steps) approaches to produce nylon 11 precursors from either ricinoleic acid or oleic acid derivatives are reported in patents8–14 and journals,15–22 but have likely not been utilized for industrial process.
While there is one study on a concise three-step metathesis-based approach for producing nylon 11 from oleic acid,17 the method has previously not been applied for producing nylons 12 or 13, likely due to challenges associated with selectivities (discussed in more detail in the Results section). The other reported methods to produce nylons 12 and 13 from non-petrochemical sources are lengthy, and also employ exotic fatty acids as starting materials. For example, there are several reported nylon 12 precursor syntheses that use ricinoleic acid from castor beans as the feedstock and require 4–6 steps.15,16,20,21,23 Synthesis of nylon 13 has not been fully studied except for one reported approach that involved several steps from either erucic or lesquerolic acid, another atypical acid available from rapeseed oil.24
Herein, we report the successful application of cross metathesis to synthesize nylon 12 and 13 precursors from oleic acid. Our method provides a shorter and simpler route for production of both nylon 12 and nylon 13 from an abundant and inexpensive, and thus preferred starting material.
Results and discussion
The previously reported cross metathesis approach to produce the nylon 11 precursor (Fig. 1(a)) involved reacting methyl oleate (1) or ricinoleate (2) with acrylonitrile.15,17,20 The resulting 10-cyano-9-decenoate (7) was subjected to high-pressure hydrogenation to remove all unsaturation and deliver the nylon 11 precursor (3) (Fig. 1, eqn (1)). Nylon 12 precursor was also prepared in analogous fashion by cross metathesis with acrylonitrile and methyl 10-undecylenate (4) prepared from pyrolysis of 2 (Fig. 1, eqn (2)). In related approaches, methyl oleate (1) or ricinoleate (2) was first converted to their derivatives such as dimers or terminal alkenes before subjecting to metathesis – these routes showed improved throughput to the original approach, although lengthened the overall synthetic steps.15,16,20,21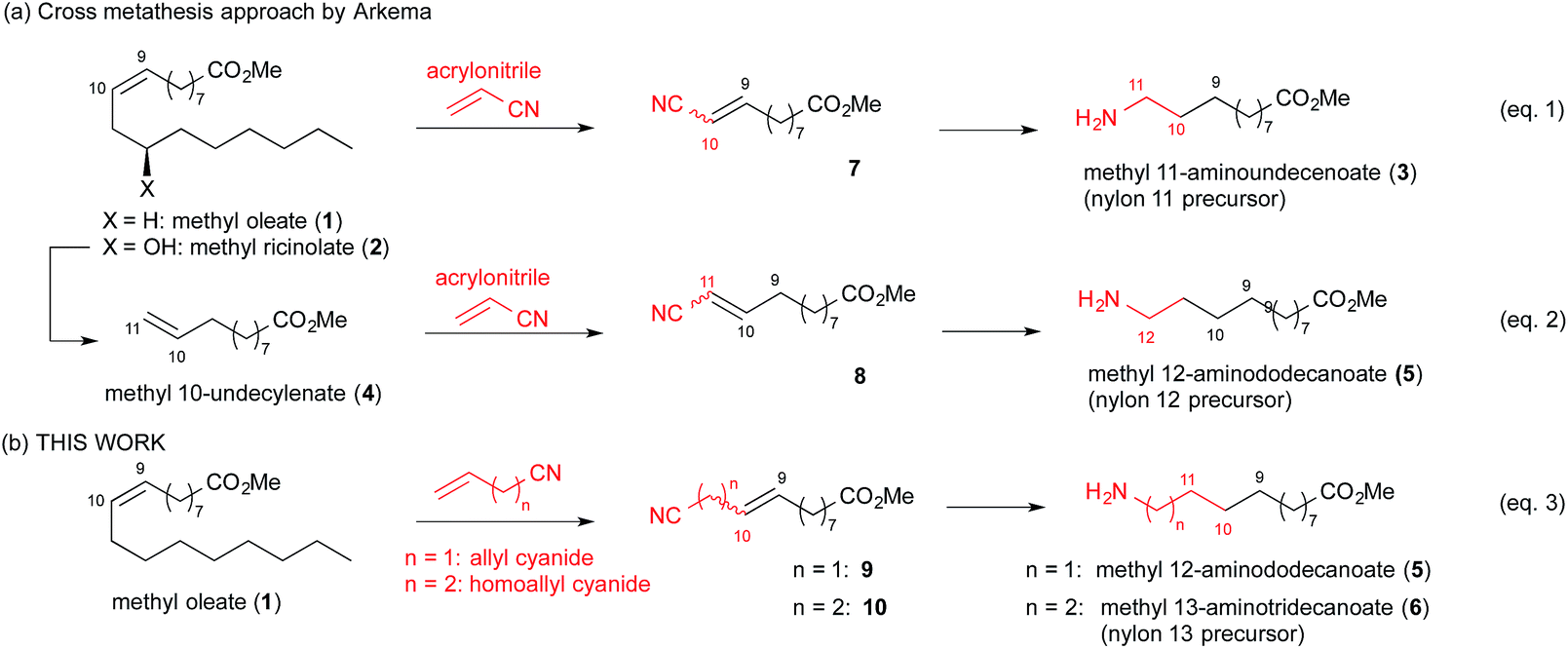 | ||
| Fig. 1 (a) Previously reported cross metathesis approach. (b) Our approach to nylon precursors from methyl oleate. | ||
Since our intention was to find a direct pathway toward nylon 12 and 13 precursors from oleic acid, we attempted cross metathesis of methyl oleate with alkenylcyanides with longer chain length (Fig. 1, eqn (3)). In the first step, we subjected methyl oleate to cross metathesis with allyl or homoallyl cyanides with the intent of generating the corresponding cyano ester intermediates 9 and 10. Subsequently, the double and triple bonds of these intermediates could be hydrogenated to deliver the nylon 12 and 13 precursors 5 and 6, respectively. Our approach, although related to the reported nylon 11 synthesis,17 behaved completely differently when their reaction conditions were used.25 In this report, we disclose our studies to overcome the issue, as well as eventual success in delivering the desired nylon precursors.
We began our investigation of the cross metathesis step using allyl cyanide and methyl 9-decenoate (11), a model substrate for methyl oleate (Table 1). Under the reported reaction conditions (entries 1–3), while the reaction conversion is good (>92%) at the temperature >95 °C, only 25–30% (by GC area) of the desired product was seen. The complex mixture of other products comprises unsaturated cyano esters with various alkyl chain lengths, with molecular weight varying by 14, which led us to speculate that they arose from olefin isomerization. Other major side-products of the reaction were series of dimers with structures yet to be determined.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Additiveh (mol%) | Catalyst (mol%) | Temperature (°C) | Time (h) | Conversion (%) | GC area% | |
| 9 | Dimers | ||||||
| a Reaction conditions: to a flask containing methyl 9-decenoate (0.1 mmol), allyl cyanide (0.5 mmol), and toluene (2 mL) was added Hoveyda–Grubbs 2nd generation catalyst (1–2 mol%) in toluene (1 mL) dropwise over 1 h.b 2 equiv. of allyl cyanide was used.c The catalyst added in one portion.d 0.5 mL of solvent was used for catalyst delivery; no other solvent was used.e 10 equiv. of allyl cyanide was used.f 12 mL (11 mL + 1 mL) of solvent for the whole reaction was used instead of 3 mL (2 mL + 1 mL).g Grubbs 2nd generation catalyst was used.h BB: benzoquinone; AA: acetic acid.i Approximate values. | |||||||
| 1 | — | 1 | 80 | 17 | 6.9 | 6.1 | 0 |
| 2b | — | 1 | 95 | 21 | 94.7 | 25.5 | 35.8 |
| 3 | — | 1 | 95 | 8 | 92.2i | 28.2 | 21.7 |
| 4 | BQ (10) | 1 | 95 | 5 | 78.7i | 52.0 | 7.8 |
| 5 | BQ (10) | 2 | 95 | 4 | 92.2 | 49.2 | 17.0 |
| 6 | AA (10) | 2 | 95 | 6 | 48.1 | 20.7 | 4.5 |
| 7 | BQ (10) | 2 | 110 | 6 | 84.4 | 56.4 | 9.0 |
| 8c | BQ (10) | 2 | 95 | 6 | 42.7 | 23.5 | 7.8 |
| 9g | BQ (10) | 2 | 95 | 6 | 49.1 | 27.8 | 3.3 |
| 10b | BQ (10) | 2 | 110 | 4 | 93.8 | 44.2 | 11.1 |
| 11d | BQ (10) | 2 | 95 | 6 | 64.6 | 39.2 | 11.0 |
| 12e | BQ (10) | 2 | 110 | 6 | 37.7 | 16.9 | 4.0 |
| 13f | BQ (50) | 2 | 110 | 4 | 84.7 | 42.2 | 3.0 |
| 14 | BQ (50) | 2 | 110 | 4 | 78.8 | 58.3 | 5.5 |

In order to suppress the undesired isomerization, the reaction was examined with several additives.22,26 We found that addition of 1,4-benzoquinone nearly completely suppressed the side reaction in our system (Table 1, entry 4). In order to fully suppress the isomerization, 50 mol% of the additive was required (entries 12 vs. 14).
Subsequently, the other reaction parameters were screened to suppress dimerization and to further improve the reaction conversion. The reaction conversion was good at temperature >95 °C; however, the best product profile was observed at 110 °C (entries 5 vs. 7). The conversion was further improved by a continuous injection of the ruthenium catalyst over 1–2 h period (entries 5 vs. 8). The reactant concentration ∼0.033 M was found to provide the best selectivity for the desired reaction (entries 11, 13 and 14). The preferred molar ratio of allyl cyanide was found to be five equivalents (entries 2 vs. 3, 10 vs. 12). Use of cross metathesis for oleochemical production from fatty acids has been a subject of several reviews.19,22,27 Consistent with other reports, Hoveyda–Grubbs second generation catalyst showed better conversion than Grubbs catalyst (entries 5 and 9) for our system. At least 2 mol% of this catalyst was required to achieve full conversion under the reaction conditions shown (entries 4 vs. 5).
It is known that solvent selection significantly influences conversion and selectivity in metathesis reactions. In particular, halogenated solvents are considered to be superior.28 In our system, solvents with lower boiling point were not suitable (Table 2, entry 2) and fluorinated solvents increased the dimer formation (entries 3 and 5). However, use of chlorobenzene provided good reaction conversion and better selectivity than other solvents (entries 4 and 6).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Time (h) | Conv. (%) | GC area% | |
| 9 | Dimers | ||||
| a Reaction condition: to the flask containing methyl oleate (0.1 mmol), allyl cyanide (0.5 mmol), 1,4-benzoquinone (0.05 mmol), in chlorobenzene (2 mL) was added Hoveyda–Grubbs 2nd generation catalyst (2 mol%) in chlorobenzene (1 mL) was added drop wise at 110 °C.b Reaction temperature: 80 °C.c 4.5 mol% of the catalyst was used.d Isolated yield. | |||||
| 1 | C6H5CH3 | 4 | 78.8 | 58.3 | 5.5 |
| 2b | (CH2Cl)2 | 8 | 41.2 | 17.8 | 11.6 |
| 3 | C6F5Cl | 2 | 95.3 | 60.1 | 20.6 |
| 4 | C6H5Cl | 2 | 90.3 | 62.6 | 13.8 |
| 5 | C6F5CF3 | 5 | 77.2 | 18.6 | 46.0 |
| 6c | C6H5Cl | 3 | 93.5 | 71.2 (58)d | 4.1 |

Subsequently, these optimized cross metathesis conditions for methyl 9-decenoate (11) were applied to methyl oleate (1) (Table 3). It was observed that dimerization was suppressed with this substrate likely due to its lower reactivity. Use of 1 instead of 11 made the product analysis more complex, because of formation of non-volatile by-products, including methyl 9-decenoate (11).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Time (h) | Timeb (h) | Conv. (%) | GC area% | ||
| 9 | Dimers | 11 | ||||
| a Reaction conditions: to the flask containing methyl oleate (0.1 mmol), allyl cyanide (0.5 mmol), 1,4-benzoquinone (0.05 mmol), in chlorobenzene (2 mL) was added Hoveyda–Grubbs 2nd generation catalyst (2 mol%) in chlorobenzene (1 mL) added dropwise at 110 °C.b Catalyst addition time.c Reaction temperature: 95 °C.d 10 equiv. of allyl cyanide was used.e 4.5 mol% of catalyst was used.f 3 mol% of catalyst was used.g GC yield (quantified).h Isolated yield. | ||||||
| 1 | 3 | 1 | 78.5 | 46.5 | 3.9 | 14.5 |
| 2c | 3 | 1 | 70.2 | 34.4 | 4.0 | 14.9 |
| 3 | 4 | 2 | 85.3 | 49.0 | 5.6 | 9.3 |
| 4 | 4 | 3 | 87.0 | 55.3 (55)h | 7.5 | 9.2 |
| 5d | 4 | 2 | 38.5 | 16.0 | 0.0 | 9.8 |
| 6e | 3 | 2 | 86.3 | 37.5 (56)g | 7.1 | 7.0 |
| 7f | 4 | 2 | 96.3 | 47.1 | 4.1 | 11.7 |

We have examined several reaction parameters in the attempt to optimize the reaction. We first examined use of lower temperature (95 °C) with hope to lower the catalyst deactivation and increase the selectivity. However, the lower temperature reactions resulted in increased formation of 11 and dimers (entry 2). To suppress the catalyst decomposition, the catalyst addition time was further extended. This attempt was partially successful and extending the time to 3 h improved the conversion as well as the selectivity for 9 (entries 3 and 4). Attempt to drive 11 to desired 9 with use of excess of cyanide led to low reaction conversion, possibly due to catalyst poisoning (entry 5). Increasing the catalyst loading up to 4.5 mol% only provided similar reaction profiles (entries 6 and 7). To date, we concluded that the current best reaction conditions for this conversion to be the one used in entry 3 or 4. This procedure reliably provides the desired 8 in ∼55% yield while suppressing olefin isomerization and dimer formation (<10%).
Cross-coupling with homoallyl cyanide was also investigated using both 9-decenoic acid (Table 4) and methyl oleate (Table 5). Some reaction tuning was required in order to achieve a good conversion with this coupling partner. In particular, we found that the reaction conversion was sensitive to equivalency of homoallyl cyanide. Under the optimal conditions used for allyl cyanide metathesis, the reaction conversion was as low as 4–5% (Table 4, entries 5 and 6) or 23% if using methyl oleate (1) (Table 5, entry 6). We suspected that this behaviour may relate to the faster dimerization of homoallyl cyanide. In any event, lowering the homoallyl cyanide equivalency to 1–2 equiv. resulted in good reaction conversion and selectivity with both substrates (Table 4, entry 2; Table 5, entry 4). Thus, both methyl 9-decenoate (11) and methyl oleate (1) effectively underwent cross-metathesis reaction with non-conjugated nitriles. We were able to obtain the desired product at a comparable isolated yield of 43% from this reaction.
|
|||||
|---|---|---|---|---|---|
| Entry | Time (h) | Cyanide (mmol) | Conv. (%) | GC area% | |
| 10 | Dimers | ||||
| a Reaction condition: 0.1 mmol of methyl oleate, 50 mol% 1,4-benzoquinone, 2 mol% of Hoveyda–Grubbs 2nd generation in chlorobenzene (1 mL) added dropwise into 1.5 mL of chlorobenzene at 110 °C. | |||||
| 1 | 6 | 0.1 | 82.2 | 43.8 | 31.8 |
| 2 | 6 | 0.15 | 83.5 | 51.2 | 27.6 |
| 3 | 4 | 0.15 | 78.0 | 48.1 | 25.0 |
| 4 | 6 | 0.25 | 78.9 | 48.9 | 24.1 |
| 5 | 6 | 0.25 | 56.3 | 38.5 | 15.9 |
| 6 | 4 | 0.5 | 4.7 | 4.0 | 0.0 |
| 7 | 6 | 0.5 | 4.2 | 4.2 | 0.6 |

|
||||||
|---|---|---|---|---|---|---|
| Entry | Time (h) | Cyanide (mmol) | Conv. (%) | GC area% | ||
| 10 | Dimers | 11 | ||||
| a Reaction condition: 0.1 mmol of methyl oleate, 50 mol% 1,4-benzoquinone, 2 mol% of Hoveyda–Grubbs 2nd generation in chlorobenzene (1 mL) added dropwise into 1.5 mL of chlorobenzene at 110 °C.b Isolated yield. | ||||||
| 1 | 6 | 0.15 | 68.6 | 31.5 | 15.3 | 7.8 |
| 2 | 6 | 0.25 | 78.8 | 42.9 | 10.9 | 10.1 |
| 3 | 8 | 0.25 | 74.4 | 34.5 | 12.6 | 9.9 |
| 4 | 4 | 0.25 | 88.7 | 52.3 (42.9)b | 8.7 | 16.2 |
| 5 | 6 | 0.3 | 77.6 | 38.0 | 7.5 | 16.8 |
| 6 | 6 | 0.5 | 23.3 | 0.4 | 6.6 | 11.7 |

In the second step, although hydrogenation of olefins is an established reaction that many catalysts are available, the reduction of nitriles to amines conventionally uses stoichiometric strong hydride reducing agents such as lithium aluminium hydride or borane, or hydrosilylation with Lewis acids such as titanium isopropoxide. Alternately, strong heterogeneous catalysts such as RANEY® nickel or cobalt can be used to catalyze addition of molecular hydrogen, but reaction conditions are harsh and only moderate selectivity is afforded. Studies on hydrogenation of nitriles with homogenous catalysts have been limited although catalysts based on rhodium, iridium, rhenium, and ruthenium have recently been investigated.29 Most recent studies used ruthenium complexes, which by far gave the best selectivity under mild reaction conditions. Many studied catalytic systems use phosphine ligands as well as potassium tert-butoxide additive. Typical reactions conditions are 80–140 °C at H2 pressure of 14–75 bars. It was also shown that milder reaction conditions could be used when the phosphine ligands of the metal complex are replaced with carbene ligands. Finally, the metathesis catalyst has also been shown to facilitate the hydrogenation reaction, and that it is possible to use residual catalyst from metathesis for hydrogenation.20,21
The previous studies for hydrogenation of fatty acid derivatives indicated that either the Grubbs or Hoveyda–Grubbs second generation catalyst would be effective.20,21 However, only Grubbs second generation catalyst provided the desired product in our experiments. Using this catalyst, we found that toluene, benzene, or chlorobenzene solvents were all suitable and the reaction temperature of 80 °C provided the best overall conversion. Thus these reaction conditions were adopted for the subsequent studies (Table 6). The base additive is essential in this catalyst system, and 30 mol% of potassium tert-butoxide was sufficient to ensure the full reaction conversion (entries 1 and 2 vs. 3–11). The catalyst loading of >2 mol% was found essential for good conversion (entries 3–5; 8–10). The reaction mixture was kept for 17–44 h under hydrogen pressure in between 20–25 bar. These reaction conditions consistently provided >50% (by GC area) of the desired methyl 12-aminododecanoate (5) (entries 4 and 6). The same reaction conditions, when used to hydrogenate C13 cyano ester (10), resulted in comparable yields (entry 6, 7 vs. 9, 10). These products were readily isolated and purified by column chromatography to yield 62% and 53% of the nylon 12 and nylon 13 precursors.
While the starting material, oleic acid, can be supplied economically from many renewable resources, our method would be particularly useful if a crude lipid extracts from microalgae could be directly used.4–6 Thus, the cross-metathesis reaction was tested using a mixture of fatty acid methyl esters (FAMEs) obtained from algal biomass by our recently developed reactive-extraction technology which enables recovery of algal lipids as FAMEs in a single step.30 We are pleased to find that both cross-metathesis with acrylonitrile and allyl cyanide proceeded smoothly consuming only unsaturated FAMEs and leaving the saturated FAMEs behind (Table 7 and Fig. 2). Even under un-optimized conditions, both reactions – with acrylonitrile (entry 1) and with allyl cyanide (entry 2) – provided the desired cyano esters 7 or 9 in close to comparable conversion but lower selectivity to control reactions (acrylonitrile: entry 3; allyl cyanide Table 3, entry 3). To the extent of our knowledge, this is the first example when cross-metathesis was demonstrated using crude algal lipid. It should be noted that throughput from use of crude algal lipid would be greater if applied to biomass that has a higher content of C9-unsaturated lipids.
 | ||
| Fig. 2 (a) A crude algal lipid containing a mixture of FAMEs. (b) Cross-metathesis with acrylonitrile. (c) Cross-metathesis with allyl cyanide. | ||
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | n | Cat. (mol%) | t-BuOK (mol%) | Pressure (bar) | Time (h) | GC area (%) | ||
| 5 or 6 | 12 or 13 | By products | ||||||
| a Reaction condition: 20 mg of methyl 11-cyano-9-undecenoate, 30 mol% of t-BuOK, 3 mol% of Grubbs 2nd generation catalyst, 3 mL of chlorobenzene at 80 °C, 30 bars during 20 h with stirring.b Extending the reaction period for additional 45 h under otherwise same conditions did not improve the product profile.c Isolated yield. | ||||||||
| 1 | 1 | 1 | 15 | 20 | 20 | 1.4 | 71.6 | 15.4 |
| 2 | 1 | 3 | 15 | 25 | 20b | 6.8 | 55.6 | 29.6 |
| 3 | 1 | 1 | 30 | 20 | 17 | 10.4 | 83.0 | 1.6 |
| 4 | 1 | 2 | 30 | 20 | 17 | 78.3 | 10.6 | 6.2 |
| 5 | 1 | 3 | 30 | 20 | 17 | 68.5 | 2.3 | 17.4 |
| 6 | 1 | 3 | 30 | 25 | 20b | 56.8 | 4.1 | 27.0 |
| 7 | 1 | 3 | 30 | 24 | 44 | 68.6 (62)c | 2.6 | 16.2 |
| 8 | 2 | 1 | 30 | 20 | 19 | 41.0 (53)c | 30.3 | 16.9 |
| 9 | 2 | 2 | 30 | 20 | 19 | 58.2 | 2.9 | 30.0 |
| 10 | 2 | 3 | 30 | 20 | 19 | 43.3 | 2.1 | 42.2 |

|
||||||
|---|---|---|---|---|---|---|
| Entry | Time (h) | Timec (h) | Conv. (%) | GC area % | ||
| 7 or 9 | Dimers | 11 | ||||
| a Reaction condition: 0.1 mmol of methyl oleate, 1 mol% of Hoveyda–Grubbs 2nd generation in toluene (1 mL) added dropwise into 1.5 mL of toluene at 95 oC.b Reaction condition: 0.1 mmol of methyl oleate, 50 mol% 1,4-benzoquinone, 2 mol% of Hoveyda–Grubbs 2nd generation in chlorobenzene (1 mL) added dropwise into 1.5 mL of chlorobenzene at 110 oC.c Catalyst addition time.d Control experiment using methyl oleate (1) and acrylonitrile under the reaction conditionsa. | ||||||
| 1 | 2 | 1 | 95.9 | 46.4 | 3.8 | 10.8 |
| 2 | 4 | 2 | 71.7 | 32.0 | 5.4 | 10.9 |
| 3d | 2.5 | 1 | >99 | 88.0 | 0 | 1.4 |

Conclusion
We have demonstrated synthesis of nylon 12 and nylon 13 precursors from methyl oleate via cross metathesis reactions with allyl and homoallyl cyanides, respectively. Our work represents the shortest synthetic route to these precursors from the abundantly available oleic acid. The key feature of our approach includes use of 1,4-benzoquinone as additive, which completely suppressed the undesired isomerization side reaction during the metathesis step. Our approach not only enables preparation of nylon 12 and 13 precursors directly from methyl oleate, but also offers new pathways to access precursors of other nylons of different chain-length, economically and sustainably. Finally, our results demonstrate that cross metathesis could be applied directly to crude fatty acid methyl ester extracts from algal biomass.Acknowledgements
This work was financially supported by National Science Foundation (CHE-1230609) and the University of Toledo (Interdisciplinary Research Initiation Program, as well as start-up funds to K. Y.).Notes and references
- R. J. Palmer, Kirk-Othmer Encyclopedia of Chemical Technology, 5th Edn, 2006, vol. 19, pp. 772–797 Search PubMed.
- A. Chauvel and G. Lefebvre, Petrochemical Processes, Editions Technip, 1989 Search PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- M. Hannon, J. Gimpel, M. Tran, B. Rasala and S. Mayfield, Biofuels, 2010, 1, 763–784 CrossRef CAS.
- D. Y. Murzin, P. Mäki-Arvela and D. A. Aranda, Biofuels, 2014, 5, 29–32 CrossRef CAS.
- T. M. Mata, A. A. Martins and N. S. Caetano, Renewable Sustainable Energy Rev., 2010, 14, 217–232 CrossRef CAS PubMed.
- A. Behr, A. Westfechtel and J. Pérez Gomes, Chem. Eng. Technol., 2008, 31, 700–714 CrossRef CAS.
- J. L. Dubois, US Pat., 8450509 B2, 2013.
- J.-L. Couturier and J.-L. Dubois, Pat., 2012–FR51771 2013030481 A1, 2013.
- J.-L. Couturier and J.-L. Dubois, Pat., 2011–57065 2978763 A1, 2013.
- J.-L. Couturier and J.-L. Dubois, Pat., 2012–FR51770 2013017782 A1, 2013.
- J.-L. Couturier, J.-L. Dubois, X. Miao, C. Fischmeister, C. Bruneau and P. Dixneuf, Pat., 2011–EP2295 2011138051, 2011.
- J.-L. Dubois and J.-L. Couturier, Pat., 2011–61757 2984309 A1, 2013.
- J.-L. Dubois and J.-L. Couturier, Pat., 2011–57542 2979342 A1, 2013.
- R. Malacea, C. Fischmeister, C. Bruneau, J.-L. Dubois, J.-L. Couturier and P. H. Dixneuf, Green Chem., 2009, 11, 152–155 RSC.
- X. Miao, C. Fischmeister, P. H. Dixneuf, C. Bruneau, J. L. Dubois and J. L. Couturier, Green Chem., 2012, 14, 2179–2183 RSC.
- X. Miao, R. Malacea, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 2911–2919 RSC.
- A. Rybak and M. A. R. Meier, Green Chem., 2007, 9, 1356–1361 RSC.
- A. Rybak and M. A. R. Meier, Green Chem., 2008, 10, 1099–1104 RSC.
- X. Miao, C. Fischmeister, C. Bruneau, P. H. Dixneuf, J.-L. Dubois and J.-L. Couturier, ChemSusChem, 2012, 5, 1410–1414 CrossRef PubMed.
- X. Miao, J. Bidange, P. H. Dixneuf, C. Fischmeister, C. Bruneau, J.-L. Dubois and J.-L. Couturier, ChemCatChem, 2012, 4, 1911–1916 CrossRef CAS.
- A. Kajetanowicz, A. Sytniczuk and K. Grela, Green Chem., 2014, 16, 1579–1585 RSC.
- J. Ternel, J.-L. Couturier, J.-L. Dubois and J.-F. Carpentier, Adv. Synth. Catal., 2013, 355, 3191–3204 CrossRef CAS.
- J. L. Greene Jr, R. E. Burks Jr and I. A. Wolff, Ind. Eng. Chem. Prod. Res. Dev., 1969, 8, 171–176 Search PubMed.
- There were attempts on analogous cross-metathesis approach to prepare the nylon 11–13 precursors using methyl 9-decenoate using pentenenitrile as a coupling partner. Their attempt to prepare the nylon 12 precursor resulted in <30% yield of the desired C12 ene-cyanide in the metathesis step: R. Gurusamy, C. Zhang and A. Gaffney, Pat., WO2013 136111 A2, 2013.
- S. H. Hong, D. P. Sanders, C. W. Lee and R. H. Grubbs, J. Am. Chem. Soc., 2005, 127, 17160–17161 CrossRef CAS PubMed.
- C. Bruneau, C. Fischmeister, X. Miao, R. Malacea and P. H. Dixneuf, Eur. J. Lipid Sci. Technol., 2010, 112, 3–9 CrossRef CAS.
- C. Samojłowicz, M. Bieniek, A. Pazio, A. Makal, K. Woźniak, A. Poater, L. Cavallo, J. Wójcik, K. Zdanowski and K. Grela, Chem.–Eur. J., 2011, 17, 12981–12993 CrossRef PubMed.
- S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 8, 289–302 CrossRef.
- S. Viamajala, D. Nelson and R. Sims, US Pat., 2011-US24549 2011100563 A2, 2011.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details of the synthetic procedures and characterization of all the new compounds. See DOI: 10.1039/c4ra10980e |
| ‡ These authors contributed equally to this study. |
§ Current address: 270 Rue du Maconnais, 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Chambery, France. 000 Chambery, France. |
| This journal is © The Royal Society of Chemistry 2014 |