
Strain hardening and highly resilient hydrogels crosslinked by chain-extended reactive pseudo-polyrotaxane†
Abstract
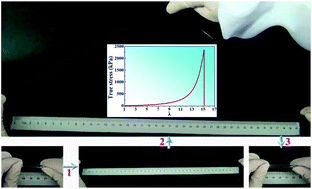
Strain hardening and high resilience are two unique mechanical characteristics of many soft biological hydrogels. However, these properties, especially the strain hardening behaviour, are generally not seen in synthetic polymer hydrogels. Here, hydrogels are prepared by free-radical copolymerization of acrylamide and chain-extended vinyl-modified pseudo-polyrotaxane, which acts as a multifunctional crosslinker. The reactive pseudo-polyrotaxane is based on a β-cyclodextrin monomer and amine-terminated PEG–PPG–PEG (Pluronic F127). The obtained hydrogels can be stretched from 10 to more than 26 times their original length before breaking and withstand a compression strain of 95% and even 98% without fracture. Tensile stretching tests show obvious strain hardening behaviours in high stretching and compression deformation regimes. The strain hardening behaviour in stretching deformation is considered to be the orientation and aggregation of the movable crosslinkers along the axial polymer backbone. Moreover, the formation of a second supramolecular network due to the chain-extension effect may also be responsible for it. Highly resilient behaviours with almost no hysteresis and residual strains are also observed even with a maximum strain of λ = 12 because of the inherent freely movable character of the crosslinkers.
 Please wait while we load your content...
Please wait while we load your content...

