
Metal-free synthesis of substituted pyridines from aldehydes and NH4OAc under air†
Rulong Yan*a,
Xiaoqiang Zhou a,
Ming Lib,
Xiaoni Lia,
Xing Kanga,
Xingxing Liua,
Xing Huoa and
Guosheng Huang*a
a,
Ming Lib,
Xiaoni Lia,
Xing Kanga,
Xingxing Liua,
Xing Huoa and
Guosheng Huang*a
aState Key Laboratory of Applied Organic Chemistry, Key Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, Department of Chemistry, Lanzhou University, Lanzhou, P. R. China. E-mail: yanrl@lzu.edu.cn; hgs@lzu.edu.cn; Fax: +86 931 8912596; Tel: +86 931 8912586
bJinchuan Group Co., Ltd., Jinchuan, P. R. China
First published on 1st October 2014
Abstract
A metal-free and efficient method for the synthesis of substituted pyridines with aldehydes and NH4OAc under mild conditions using air as the oxidant was developed. This oxidative cyclization process involves direct C–H bond functionalization, C–C/C–N bond formation and C–C bond cleavage.
Substituted pyridines as one of the most prevalent heterocyclic compounds are important building blocks for many natural products, bioactive molecules, and functional materials,1 For example, many herbicides and fungicides as well as thousands of drugs contain the skeleton of pyridines.2 Thus, they have drawn considerable interest for synthetic chemists. Accordingly, numerous of well-documented traditional and modern methods have been developed for the construction of pyridines and their derivatives, among which the tradition metal catalyzed cycloaddition reactions represent as typical method for pyridines synthesis.3 Especially, the group of Eliel had reported the synthesis of substituted pyridines from aldehyde and ammonia gas via abnormal Chichibabin reaction.4 Very recently, Yoshikai group has developed syntheses of pyridines from oximes and enals (Scheme 1).5 However, most of those methods suffer from several disadvantages such as use of highly toxic metal compounds, instability of the substrates, poor yields and harsh reaction conditions. Therefore, the development of an alternative metal-free approach for the synthesis of pyridines under air remains highly desirable.
 | ||
| Scheme 1 Aldehydes and anilines in the synthesis of pyridines. | ||
Recently, the C–H activation and C–C/C–N bond formation have presented an attractive and powerful strategies for generation heteroaromatic compounds.6 In view of green and sustainable chemistry, development of economical and environmentally benign strategies for the construction of useful heterocyclic skeletons with simple and readily accessible substrates is an attractive goal in contemporary organic synthesis. Particularly, air has emerged as an ideal oxidant for the synthesis of heterocyclic compounds in a step and atom-economical fashion for its abundance, environment-friendly and numerous advantages in industry.7 During our investigation the synthesis of heterocyclic compounds using dioxygen as the oxidant,8 we discovered a rather surprising formation of substituted pyridines from 2-phenylacetaldehydes and ammonium acetate under air.
In our initial experiments, 2-phenylacetaldehyde (1a) and NH4OAc (2) were chosen as the model substrates for the reaction, as shown in Table 1. Treating the substrate 1a and 2 in DMSO at 120 °C, to our delight, an interesting product 3,5-diphenylpyridine (3a) was obtained in 72% yield, and we confirmed the structure of 3a unambiguously through an X-ray crystal analysis. Among the N-source we examined, NH4OAc was found the best substrate for the reaction (Table 1, entries 1–6). Further studies showed that NaHCO3 was the most efficient additive for the reaction when DMSO was used as solvent, affording the desired product in 76% yield (Table 1, entries 7–10). After screening on different parameters, the highest yield of 3a was achieved, when the reaction was carried out at 90 °C in 1,4-dioxane (Table 1, entry 14).
|
|||||
|---|---|---|---|---|---|
| Entry | N source | Additive | Solvent | Temp | Yieldb |
| a Reaction conditions: 1a (0.3 mmol), 2 (0.9 mmol), additive (0.3 mmol), solvent (1 mL), 5 h.b Yields of isolated products.c The reaction was carried out under O2 (1 atm). | |||||
| 1 | NH4OAc | DMSO | 120 | 72 | |
| 2 | NH4HCO3 | DMSO | 120 | 68 | |
| 3 | NH4Cl | DMSO | 120 | 42 | |
| 4 | NH3·H2O | DMSO | 120 | 75 | |
| 5 | (NH4)2C2O4 | DMSO | 120 | 66 | |
| 6 | NH2OH·HCl | DMSO | 120 | 0 | |
| 7 | NH4OAc | K2CO3 | DMSO | 120 | 72 |
| 8 | NH4OAc | NaHCO3 | DMSO | 120 | 76 |
| 9 | NH4OAc | NaOAc | DMSO | 120 | 69 |
| 10 | NH4OAc | HOAc | DMSO | 120 | 44 |
| 11 | NH4OAc | NaHCO3 | DMF | 120 | 75 |
| 12 | NH4OAc | NaHCO3 | PhMe | 100 | 58 |
| 13 | NH4OAc | NaHCO3 | EtOH | 70 | 67 |
| 14 | NH4OAc | NaHCO3 | 1,4-Dioxane | 90 | 80 |
| 15c | NH4OAc | NaHCO3 | 1,4-Dioxane | 90 | 63 |
| 16 | NH4OAc | NaHCO3 | H2O | 90 | 21 |

With the optimized reaction conditions in hand, we explored the substrate scope of this reaction, and the results are illustrated in Table 2. Generally, the reaction of substituted aldehydes and NH4OAc proceeded smoothly and afforded the corresponding substituted pyridines with high efficiency (Table 2). It is observed that the nature of the substituent on the aromatic rings did not significantly affect the efficiency on the yields of the products. The ortho-, meta-, and para-substituted alkyl groups, as well as the electron-donating and electron-withdrawing groups were well tolerated, such as methyl, methoxyl, fluoro groups (3a–3n). The 3,5-di(naphthalen-1-yl)pyridine 3m was obtained in 61% yield when 2-(naphthalen-2-yl)acetaldehyde 1m was employed as the substrate. Moreover, when 2-(furan-2-yl) acetaldehyde was subjected to the transformation, the desired product also was obtained in 48% yield (3o).
|
|---|
| a All the reaction were carried out in the presence of 1 (0.3 mmol), 2 (0.9 mmol) and NaHCO3 (0.3 mmol) in 1 mL 1,4-dioxane at 90 °C for 5 h. |
 |

Moreover, different aldehydes also can work together under the optimized conditions, and the scope was further expanded (Scheme 2). The reaction of 1b with 1g afforded 3p in 34% yield, and 1b with 1i afforded 3q in 36% yield, respectively. Meanwhile, the 3b, 3g and 3i were also detected in this transformation.
 | ||
| Scheme 2 Diversity of polysubstituted pyridines synthesis. | ||
Further experiments were conducted for the reaction of substituted 3-phenylpropanal and NH4OAc under optimized conditions. As shown in Table 3, the nature of substituted groups on 3-phenylpropanals can not significantly affect this transformation. The substrates with electron-donating and electron-drawing group all can proceed well under the optimized conditions and give the desired products in moderate yields (4a–4g). However, the reaction did not work when the butyraldehyde were employed as the reaction substrates (4k).
|
|---|
 |

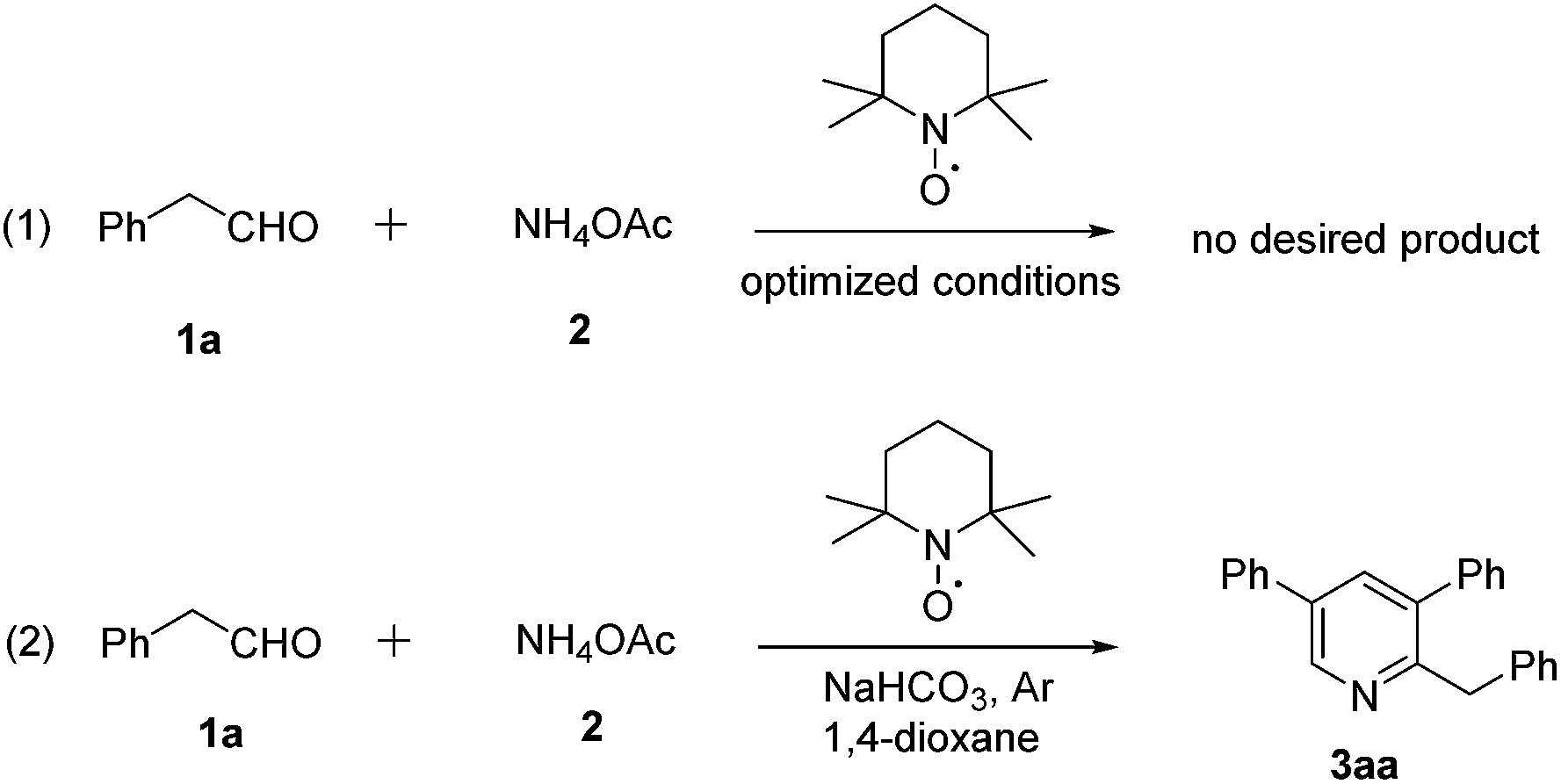
To probe the mechanism further, some experiments were investigated. Firstly, annulation of 1a and 2 were carried out under standard conditions, benzaldehyde was detected by GC-MS in the reaction system. The radical trapping experiment was also performed in the presence of 2,2,6,6-teramethylpiperidine oxide (TEMPO). Indeed, the addition of 2.0 equiv. of TEMPO that led to the oxidative process was remarkly suppressed and no desired product and useful intermediate was isolated (Scheme 3, step 1). Fortunately, When 2.0 equiv. of TEMPO was employed in this transformation via standard condition under argon, an unexpected compound 2-benzyl-3,5-diphenylpyridine (3aa) was detected (Scheme 3, step 2). The structure of product 3aa and 4 mean that the carbon atom of C-2 position in pyridine ring comes from the carbonyl of aldehydes in this transformation. Moreover, the product of 3aa indicates that 12 would be the intermediate of the transformation.
 | ||
| Scheme 3 Control experiments. | ||
On the basis of the results described above, a plausible mechanism with two paths is proposed in Scheme 4. First, the acetaldehyde 1 condenses with 2 to form imine 5, which would proceed aldol condensation with 1 to afford 7. The intermediate 7 equilibrates to generate enamine 8 easily.9 Then, intermediate 12 is formed via intramolecular nucleophile addition from imine 9, which is generated by the reaction of enamine 8 and 1 (Path A). Alternatively, the imine 5 also can equilibrate to generate the enamine 6. Sequently, the intermediate 10 is formed by the reaction of enamine 6 and 1. Intermolecular nucleophile addition of 10 and 1 gives rise to 11 (Path B). Then, 12 is generated by the intramolecular nucleophile. Furthermore, when substituted 3-phenylpropanals were served as substrates, the hydroperoxide 12 was converted to product 4 via the hydride elimination directly. When substituted acetaldehyde were served as substrates, the hydroperoxide 13 is provided by the combination of the intermediate 12 and O2. Moreover, the radical 14 and hydroxide radical (˙OH) are generated by decomposition of the hydroperoxide 13. The single electron transfer of 14 forms the radical 15 and aldehyde with C–C cleavage. Finally, the pyridine 3 is afforded by the radical hydride elimination of 15.
 | ||
| Scheme 4 Proposed mechanism of the synthesis pyridines. | ||
In conclusion, we have developed a simple and efficient method for the synthesis of substituted pyridines. This method constructs the skeleton of pyridine with aldehydes and NH4OAc by direct C–H functionalization, C–C/C–N bond formation and C–C bond cleavage under mild reaction conditions. The procedure, using air as oxidative agent, is a very practical, economical, and environmentally friendly protocol for the synthesis of substituted pyridines. This work was supported by National Natural Science Foundation of China (21202067) and the Fundamental Research Funds for the Central Universities (lzujbky-2014-71).
Notes and references
- (a) J. A. Joule and K. Mills, in Heterocyclic Chemistry, Blackwell, Oxford, UK, 4th edn, 2000 Search PubMed; (b) J. P. Michael, Nat. Prod. Rep., 2005, 22, 627 RSC; (c) M. Abass, Heterocycles, 2005, 65, 901 CrossRef CAS PubMed; (d) V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745 CrossRef CAS PubMed; (e) H. Sasabe and J. Kido, J. Mater. Chem., 2011, 23, 621 CrossRef CAS; (f) S.-J. Su, T. Chiba, T. Takeda and J. Kido, Adv. Mater., 2008, 20, 2125 CrossRef CAS.
- (a) G. D. Henry, Tetrahedron, 2004, 60, 6043 CrossRef CAS PubMed; (b) X. T. Tian, B. J. Qin, H. Lu, W. H. Lai, S. B. Jiang, K. H. Lee, C. H. Chen and L. Xie, Bioorg. Med. Chem. Lett., 2009, 19, 5482 CrossRef CAS PubMed.
- For selected recent achievements on pyridine synthesis, see: (a) A. Winter and N. Risch, Synthesis, 2003, 2667 CAS; (b) Y. F. Wang and S. Chiba, J. Am. Chem. Soc., 2009, 131, 12570 CrossRef CAS PubMed; (c) I. Nakamura, D. Zhang and M. Terada, J. Am. Chem. Soc., 2010, 132, 7884 CrossRef CAS PubMed; (d) M. Ohashi, I. Takeda, M. Ikawa and S. Ogoshi, J. Am. Chem. Soc., 2011, 133, 18018 CrossRef CAS PubMed; (e) C. Wang, X. Li, F. Wu and B. Wan, Angew. Chem., Int. Ed., 2011, 50, 7162 CrossRef CAS PubMed; (f) Q. Wang, C. Wan, Y. Yang, J. Zhang, L. Gao and Z. Wang, Green Chem., 2011, 13, 578 RSC; (g) S. Yamamoto, K. Okamoto, M. Murakoso, Y. Kuninobu and K. Takai, Org. Lett., 2012, 14, 3182 CrossRef CAS PubMed; (h) W. Gati, M. M. Rammah, M. B. Rammah, F. Couty and G. Evano, J. Am. Chem. Soc., 2012, 134, 9078 CrossRef CAS PubMed; (i) H. Huang, X. Ji, W. Wu, L. Huang and H. Jiang, J. Org. Chem., 2013, 78, 3374 CrossRef PubMed; (j) Z. Shi and T.-P. Loh, Angew. Chem., Int. Ed., 2013, 52, 8584 CrossRef CAS PubMed; (k) S. Michlik and R. Kempe, Angew. Chem., Int. Ed., 2013, 52, 6326 CrossRef CAS PubMed.
- E. L. Eliel, R. T. Mabride and S. Kaufmann, J. Am. Chem. Soc., 1953, 75, 4291 CrossRef CAS.
- Y. Wei and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 3756 CrossRef CAS PubMed.
- (a) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (b) C. Copret, Chem. Rev., 2010, 110, 656 CrossRef PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (e) F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977 CrossRef CAS PubMed; (f) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed.
- For some reviews, see: (a) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef CAS PubMed; (b) S. S. Stahl, Angew. Chem., 2004, 116, 3480 (Angew. Chem., Int. Ed., 2004, 43, 3400) CrossRef; (c) M. S. Sigman and D. R. Jensen, Acc. Chem. Res., 2006, 39, 221 CrossRef CAS PubMed; (d) K. M. Gligorich and M. S. Sigman, Angew. Chem., 2006, 118, 6764 (Angew. Chem., Int. Ed., 2006, 45, 6612) CrossRef; (e) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., 2011, 123, 11256 (Angew. Chem., Int. Ed., 2011, 50, 11062) CrossRef; (f) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (g) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC.
- (a) R. L. Yan, J. Huang, J. Luo, P. Wen, G. S. Huang and Y. M. Liang, Synlett, 2010, 1071 CAS; (b) R. L. Yan, J. Luo, C. X. Wang, C. W. Ma, G. S. Huang and Y. M. Liang, J. Org. Chem., 2010, 75, 5395 CrossRef CAS PubMed; (c) R. L. Yan, H. Yan, C. Ma, Z. Y. Ren, X. A. Gao, G. S. Huang and Y. M. Liang, J. Org. Chem., 2012, 77, 2024 CrossRef CAS PubMed; (d) H. Yan, R. Yan, S. Yang, X. Gao, Y. Wang, G. Hang and Y. Liang, Chem.–Asian J., 2013, 19, 4271 Search PubMed; (e) R. Yan, X. Liu, C. Pan, X. Zhou, X. Li, X. Kang and G. Huang, Org. Lett., 2013, 15, 4876 CrossRef CAS PubMed; (f) R. Yan, X. Kang, X. Zhou, X. Li, X. Liu, L. Xiang, Y. Li and G. Huang, J. Org. Chem., 2014, 79, 465 CrossRef CAS PubMed.
- N. El-Hamdouni, X. Companyo, R. Rios and A. Moyano, Chem.–Eur. J., 2010, 16, 1142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic, analytical and X-ray data. CCDC 958750. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra10880a |
| This journal is © The Royal Society of Chemistry 2014 |