
A carboxylic acid-functionalized coumarin-hemicyanine fluorescent dye and its application to construct a fluorescent probe for selective detection of cysteine over homocysteine and glutathione†
Jing Liu‡
a,
Yuan-Qiang Sun‡a,
Hongxing Zhanga,
Yingying Huoa,
Yawei Shib,
Heping Shia and
Wei Guo*a
aSchool of Chemistry and Chemical Engineering, Shanxi University, Taiyuan 030006, China. E-mail: guow@sxu.edu.cn
bInstitute of Biotechnology, Shanxi University, Taiyuan 030006, China
First published on 11th November 2014
Abstract
A carboxylic acid-functionalized coumarin-hemicyanine near-infrared (NIR) dye 1 was exploited, which possesses good water solubility (more than 50 μM) and favorable photophysical properties, especially a large Stokes shift (around 90 nm), and has been proved to be a suitable imaging agent for targeting mitochondria. With the dye platform, fluorescent probe 2, a thioester derivative of 1, was constructed for biothiols. Probe 2 can react with cysteine (Cys) via the native-chemical-ligation (NCL) and cyclization cascade reactions to lead to coumarin 2-Cys. However, the reaction of 2 with homocysteine (Hcy) or glutathione (GSH) only stays at the stage of the initial transthioesterification reaction, producing coumarin-hemicyanines 2-Hcy or 2-GSH, due to an electrostatic interaction in 2-Hcy and an unstable macrocyclic transition state in 2-GSH, both inhibiting their subsequent S,N-acyl shift. Given the distinct photophysical properties between 2-Cys and 2-Hcy (or 2-GSH), probe 2 could highly selectively discriminate Cys from Hcy/GSH. Even in the presence of Hcy or GSH, probe 2 still works well for Cys due to the reversible transthioesterification and the irreversible S,N-acyl shift in the NCL reaction. The cell imaging assays revealed that probe 2 is cell permeable and could selectively image Cys in living cells.
Introduction
Because the fluorescent indicators for calcium ion were reported by Tsien in the early 1980s,1 fluorescent probes have been recognized as efficient molecular tools that can help monitor and visualize trace amounts of samples in live cells or tissues because of their high sensitivity and high spatiotemporal resolution.2 In particular, the exploitation of reaction-based fluorescent probes has attracted increasing attention in recent years3 and has become an active research field due to their higher selectivity with large spectroscopic changes than those based on non-covalent interactions in most cases. Usually, the construction of a reaction-based fluorescent probe involves two integrated components: one is the signaling fluorophore and the other is the receptor that possesses a reaction-based binding capability. When a guest species is bound to the receptor, the photophysical characteristics of the fluorophore will change via different mechanisms. Photoinduced electron transfer (PET) is a widely used fluorescence-modulated mechanism to achieve the goal, through which the fluorescence of a fluorophore is quenched by the electron transfer from the donor to the acceptor upon excitation, and could be recovered via the inhibition of this process by guest binding. However, in practical terms, it is still difficult to accurately predicate the analytical performance of a PET-based fluorescent probe, and in many cases, the undesired background fluorescence still remains, which is especially disadvantageous for biological research. Moreover, it is more serious when near-infrared (NIR) fluorophores are used because most NIR fluorophores have relatively high-lying HOMO energy, such that the Off–On switching of NIR fluorescence via PET is less efficient.4An attractive solution to this issue is employing the strategy of the change in the π-conjugated system of a dye induced by chemical reactions.5,6 The most typical examples are the rhodamine (a carboxylic acid-functionalized xanthene dye)-based fluorescent probes. The spirocyclic form of these probes is nonfluorescent due to the less π-conjugation state; however, its ring-open form is highly fluorescent as a result of π-conjugation recovery, thereby enabling the fluorescence Off–On response with almost negligible background fluorescence. Thus, since the first rhodamine-based fluorescent probe for Cu2+ was reported by Czarnik in 1997,6a rhodamine has been regarded as a versatile platform for fluorescent probe development.6b–e However, the absorption and emission wavelengths of most rhodamine derivatives are below 600 nm. In fact, it is well established that fluorescent dyes operating in the far-red to NIR region have many advantages for biological applications, including low phototoxicity, low autofluorescence, and good tissue penetration.7 As such, some excellent rhodamine-inspired long-wavelength fluorescent dyes have been actively developed in recent years,8 such as Si-rhodamines (SiR),9 as well as the carboxylic acid-functionalized SiR,10 merocyanine dyes (CS NIR)11 and hybrids of coumarin and rhodamine12 reported by Nagano, Wu, Lin, and Wang, respectively. These dyes not only enrich the family of NIR dyes for various biological applications, but also provide the important platforms for the development of various fluorescent probes based on the spirocyclization-induced fluorescence switching mechanism. However, one of the main drawbacks of these dyes is their relatively small Stokes shifts. The small Stokes shift is often detrimental for practical applications either due to reduced emission intensity by self-absorption and inner filter effect or fluorescence detection errors because of excitation backscattering effects. Therefore, there is a strong interest in the development of the novel NIR dyes aimed at improving the spectral parameter while retaining the rhodamine-like fluorescence switching mechanism for fluorescent probe design.
In this paper, we presented a carboxylic acid-functionalized coumarin-hemicyanine fluorescent dye 1 (Scheme 1B) by combining the advantages of the large Stokes shift of coumarin dye, NIR emission of cyanine dye, and carboxylic acid functional group of rhodamine dye (Scheme 1A). In addition, besides the high molar extinction coefficient, good fluorescence quantum yield, and sufficient photostability, several striking advantages of the dye also include: (1) the fluorescence of the dye falls into the red region, thus being preferable for in vivo bioimaging; (2) the Stokes shift of the dye is obviously larger than those classical dyes, such as rhodamine, fluorescein, BODIPY, and cyanine; (3) the carboxylic acid group not only increases the hydrophilicity of the dye, but also is amenable to modification in order to construct various fluorescent probes; (4) due to its positive charge delocalized through resonance, just like the mitochondria-selective probes rhodamine 123 and tetramethylrosamine,13 the dye was found to be a suitable imaging agent for targeting mitochondria.
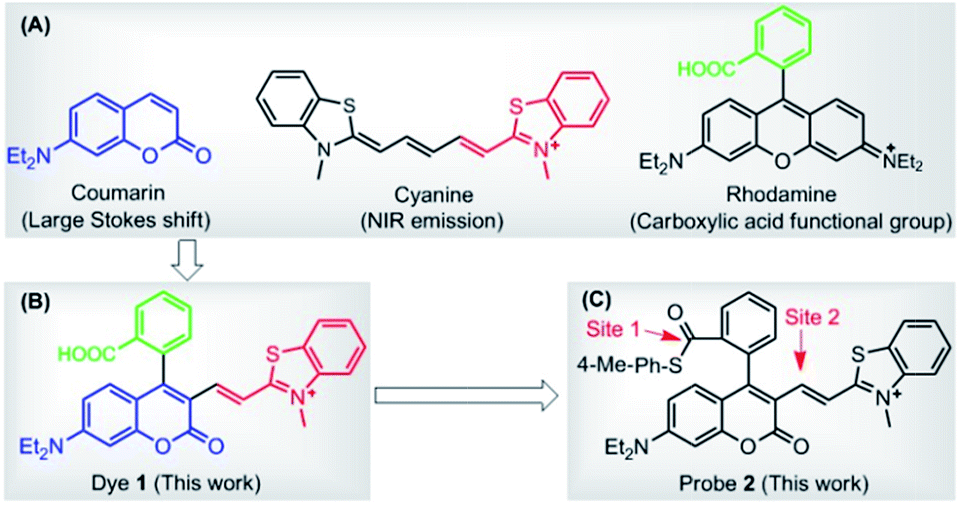 | ||
| Scheme 1 (A) Chemical structures of coumarin, cyanine, and rhodamine dyes. (B) Carboxylic acid-functionalized coumarin-hemicyanine dye 1. (C) Probe 2 based on dye 1 platform. | ||
Further, with the dye 1 platform, we synthesized a fluorescent probe 2 (a thioester of 1) (Scheme 1C) for cysteine (Cys), inspired by the widely used native chemical ligation (NCL) reaction in peptide synthesis,14 i.e. the Cys residue-induced transthioesterification and the subsequent intramolecular S,N-acyl shift (Scheme 2A). As shown in Scheme 2B, probe 2 can undergo a NCL-cyclization cascade reaction with Cys to lead to coumarin 2-Cys; however, homocysteine (Hcy) and glutathione (GSH) only trigger a transthioesterification reaction to produce coumarin-hemicyanine 2-Hcy and 2-GSH, respectively, due to a strong electrostatic interaction between 2-Hcy and an unstable macrocyclic transition state in 2-GSH, both inhibiting their subsequent S,N-acyl shift. In view of the distinct emission between 2-Cys and 2-Hcy (or 2-GSH), probe 2 could highly selectively discriminate Cys from Hcy/GSH. Notably, even in the presence of Hcy and GSH, probe 2 still works well for Cys due to the reversible transthioesterification reaction and the irreversible S,N-acyl shift.14 Assisted by laser scanning confocal microscope, we further demonstrated the potential of 2 to selectively monitor Cys in living cells. Given that Cys plays crucial roles in many physiological processes,15 and is closely related to many diseases,16 the results reported herein are encouraging and may provide an opportunity for studying the Cys-related physiological processes and diseases in biological systems.
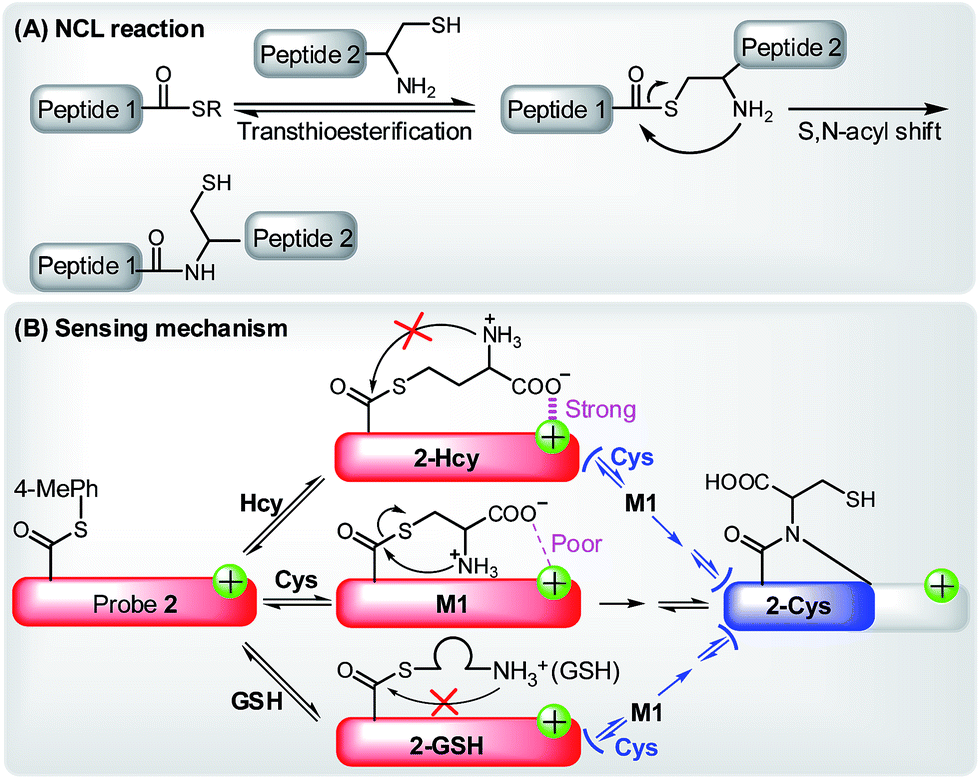 | ||
| Scheme 2 (A) The principle of NCL reaction. (B) Selectively sensing of Cys by 2 based on the NCL-cyclization reaction in the absence or presence of Hcy and GSH. | ||
Results and discussion
Synthesis and spectra properties of dye 1 and its application to specifically target mitochondria
Dye 1 was synthesized by a simple four-step procedure (Scheme 3). The starting material S4 was prepared according to the reported procedure.17 Condensation of compound S4 with diethyl malonate in acidic conditions afforded the intermediate S3, which was then hydrolyzed in the mixture of concentrated HCl and glacial acetic acid to obtain the intermediate S2. The intermediate S1 was synthesized via Duff reaction of S2. Condensation of S1 with 2,3-dimethylbenzothiazolium iodide in refluxing ethanol afforded the desired product 1 as a dark green solid. The chemical structure of 1 was confirmed by 1H NMR, 13C NMR, and HRMS spectra (ESI†). | ||
| Scheme 3 Synthesis of dye 1. | ||
The photophysical properties of dye 1 were subsequently determined with EtOH as a representative solvent. In EtOH, 1 displayed a high extinction coefficient (ε = 2.64 × 104 M−1 cm−1) with absorption maximum at 550 nm, good fluorescence quantum yield [Φ = 0.195 with cresyl violet (Φ = 0.54) as reference], red emission at 644 nm, and significant Stokes shift up to 94 nm (Fig. 1A). Such a big Stokes shift can avoid not only self-quenching by self-absorption and inner filter effect but also fluorescence detection errors due to excitation backscattering effects, and thus is very advantageous for practical application. In this regard, dye 1 appears to be preferable to those classical dyes, such as rhodamine, fluorescein, BODIPY, and cyanine (generally less than 30 nm). In addition, the photostability of 1 in EtOH was also evaluated by continuous irradiation of the dye using a 150 W Xe lamp as the light source at a 10 nm slit width at the maximal absorption wavelength of 1. After 60 min continuous irradiation, about 98.5% of the initial fluorescence intensity was retained (Fig. 1B), indicating that the dye has sufficient photostability for biological applications.
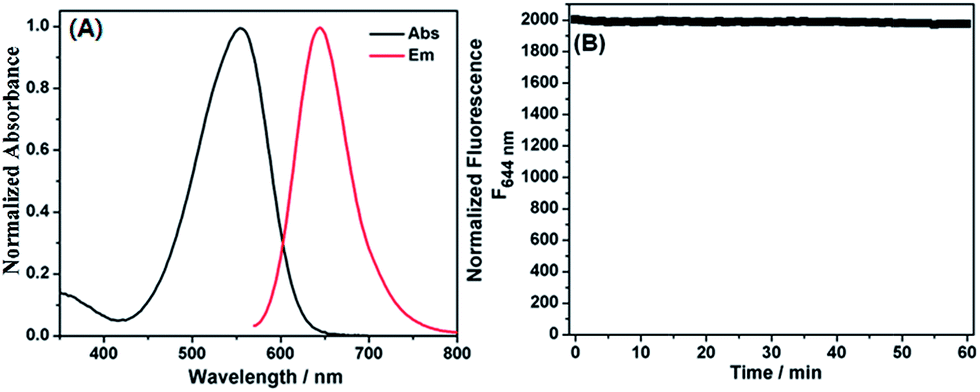 | ||
| Fig. 1 (A) Normalized absorption and emission spectra of 1 in EtOH. (B) Photostability of 1 in EtOH. The sample was continuously irradiated by a xenon lamp (150 W) at 10 nm slit width at the maximal absorption wavelength of 1. | ||
Mitochondria are the important energy-producing compartment in cells, and play crucial roles in numerous vital cellular processes. Mitochondria are also involved in various pathologies, such as Alzheimer's disease, cancer, and diabetes.18 Thus, mitochondria-staining reagents are important in biomedical research and diagnostic applications.19 Although various mitochondrial-targeting fluorescence dyes are commercially available, only a few are in the long-wavelength range. Moreover, these dyes commonly suffer from some practical limitations, such as small Stokes shift and poor photostability. It is noteworthy that dye 1 possesses a positive charge delocalized through resonance as the mitochondria-selective probes rhodamine 123 and tetramethylrosamine,13 and thus was highly expected to be a suitable imaging agent for targeting mitochondria by electrostatic interactions due to the highly negative mitochondrial membrane potentials (about −180 mV).20 Subsequently, we performed confocal fluorescence microscopy observations after co-staining the cells with a mitochondria-specific fluorescent marker MitoTracker Green FM. B16 cells or HeLa cells were seeded to a glass bottomed dish in a static overnight culture. Adhered cells were stained with dye 1 (4 μM) and MitoTracker green (0.15 μM) for 20 min each. After washing, cells were successively observed with confocal fluorescence microscopy. As shown in Fig. 2, the fine merged image in the co-staining experiments clearly confirmed that the dye could efficiently penetrate the cell membrane and specifically label mitochondria, revealing its potential for imaging and therapeutic applications. The minimal cytotoxicity of 1 was also demonstrated by MTT assay (Fig. S1, ESI†).
 | ||
| Fig. 2 Confocal fluorescence microscopy images of B16 cells (A1–D1) and HeLa cells (A2–D2) co-stained by MitoTracker Green FM (0.15 μM, 20 min) and 1 (4 μM, 20 min). (A1 and A2) image from band path of 500–550 nm upon excitation of MitoTracker Green FM at 488 nm; (B1 and B2) image from band path of 590–750 nm upon excitation of 1 at 543 nm; (C1 and C2) the corresponding overlap images; (D1,D2) the corresponding bright field images. Scale bar: 10 μm. | ||
To probe the possible spirocyclization mechanism, we subsequently investigated the absorption spectra changes of 1 in response to different pHs. Dye 1 was found to be considerably water soluble (>50 μM in PB buffer) (Fig. S2, ESI†), and showed its main absorption around 543 nm (Fig. 3A), a typical emission of coumarin-hemicyanine dye, in a wide pH range of 2–12, indicating that 1 mainly exists as the ring-open form in these cases. Similarly, in MeOH, EtOH, CH3CN, CH2Cl2, CHCl3 and PB (pH 7.4, 20 mM), 1 also exists as the ring-open form in terms of the main absorptions in the range from 540 to 580 nm (Fig. 3B). Thus, unlike those reported carboxylic acid-functionalized dyes,6,10–12 it is difficult for 1 to undergo the spirocyclization reaction presumably due to the poor reactivity of coumarin 4-position. However, when the carboxylic acid group was replaced by a more nucleophilic amide group, a cyclization reaction rather than the spirocyclization reaction could be clearly observed (see below), providing the possibility for designing fluorescent probes based on the change of π-conjugated systems.
 | ||
| Fig. 3 (A) Absorption spectra of 1 (2 μM) in the different pH conditions. (B) Absorption spectra of 1 (2 μM) in different solvents. | ||
Ratiometric fluorescent probe 2 for Cys based on dye 1 platform
Because biothiols, such as Cys, Hcy, and GSH, play crucial roles in many physiological processes, and are closely related to many diseases, a large number of reaction-based fluorescent probes have been developed in recent years to detect and sense these important species.21 However, most of these probes cannot distinguish these biothiols from each other due to their similar structure and reactivity. Because Cys, Hcy, and GSH levels are related to different physiological processes and diseases, the development of fluorescent probes that could discriminate between them is highly valuable for better understanding of their respective molecular mechanism of action. Toward this end, in addition to some important progress on Hcy22 or GSH23 fluorescent probes, unceasing efforts have been made towards Cys as follows: based on the cyclization of Cys/Hcy with aldehydes24a or acrylates,24b pioneered by the Strongin group, the selective detection of Cys/Hcy over GSH has been realized.25 Furthermore, some specific probes for Cys were developed based on either the extended version of the two strategies26,27 or Michael addition combined with steric, electrostatic and hydrogen-bonding interactions28 or the Cys-induced SNAr substitution–rearrangement reaction.29 In fact, from a design point of view, it appears to be relatively easy to preclude the interference of GSH with these strategies. However, the discrimination between Cys and Hcy still remains a hit-or-miss proposition, often a matter of luck as these two amino acids differ only by one methylene unit in their side chains.Recently, inspired by the Cys-induced NCL reaction,14,30 Strongin and Yang et al. exploited two new fluorescent probes for biothiols based on the carboxylic acid-functionalized benzothiazole-rhodol and coumarin-benzopyrylium dye platforms, respectively.31 Because the sulfhydryl and the amino groups of Cys/Hcy are both involved in the NCL reaction, the two probes show high selectivity toward Cys/Hcy over GSH, as well as other amino acids. However, they could not distinguish between Cys and Hcy due to the same reaction mechanism. Herein, by functionalizing the carboxylic acid group of dye 1, we designed and synthesized fluorescent probe 2 (Scheme 4), a thioester derivative of 1, with the expectation of improving the selectivity toward biothiols based on the NCL reaction coupled with steric or electrostatic interaction. The chemical structure of 2 was confirmed by 1H NMR, 13C NMR, and HRMS spectra (ESI†).
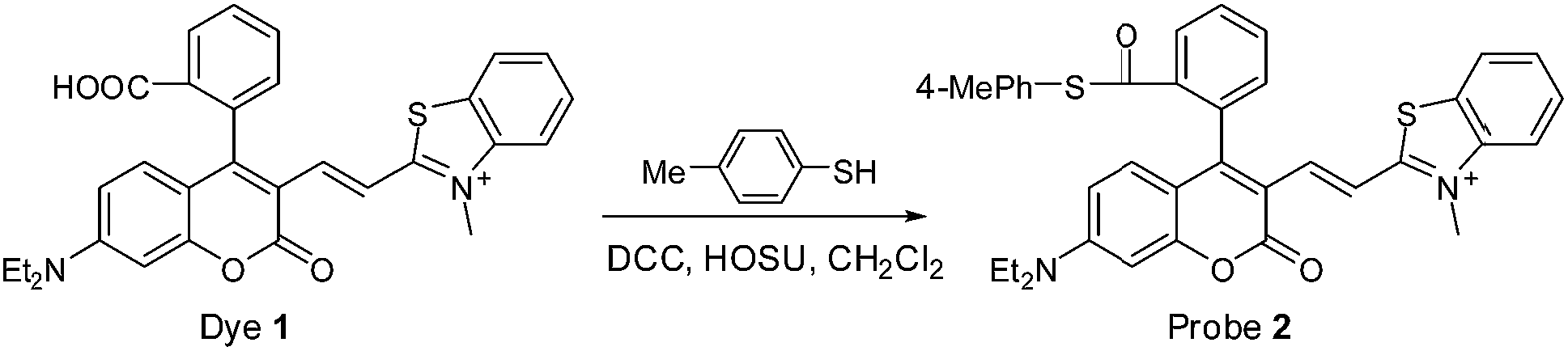 | ||
| Scheme 4 Synthesis of probe 2. | ||
With the probe in hand, we first examined its reactivity towards Cys, Hcy, and GSH through time-dependent UV-vis spectra in PB (pH 7.4, 20 mM, containing 10% DMF) at 37 °C (Fig. 4). As a model compound of amino-free thiol, N-acetylcysteine (NAC) was also tested. The free 2 showed a main absorption at 550 nm ascribed to the ring-open coumarin-hemicyanine dye. Upon addition of 20 equiv. of Cys, the initial absorption peak gradually decreased along with the simultaneous emergence of the new blue-shifted peaks at 405 nm (Fig. 4A). Such a big blue shift in absorption wavelength indicated that the reaction breaks the π-conjugation of the coumarin-hemicyanine unit in 2. Surprisingly, the addition of 20 equiv. of Hcy resulted in only a slight decrease in absorption at 550 nm along with an almost negligible increase at 405 nm (Fig. 4B). A similar case was also observed when GSH or NAC was used (Fig. 4C and D). The distinct absorption spectra changes of 2 treated with Cys and Hcy (or GSH or NAC) indicated that these reactions must go through different routes.
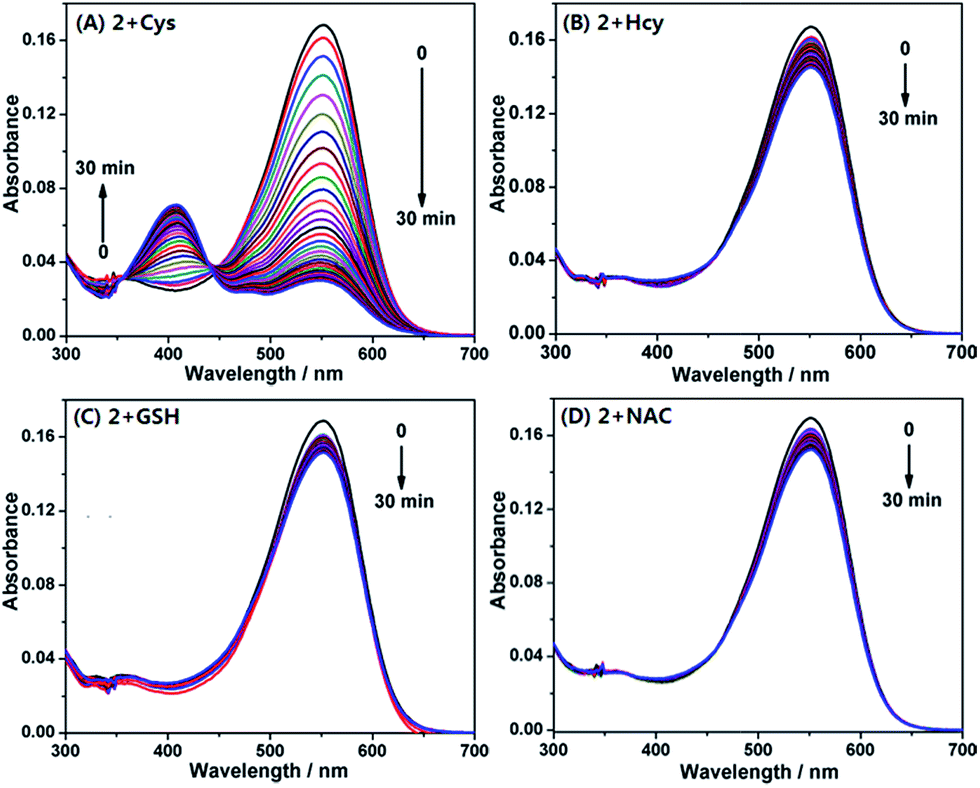 | ||
| Fig. 4 Time-dependent absorption spectra of 2 (4 μM) upon addition of 20 equiv. of Cys (A), Hcy (B), GSH (C), and NAC (D), respectively, in PB (pH 7.4, 20 mM, containing 10% DMF) at 37 °C. | ||
On the basis of the above observations, we proposed the possible reaction mechanisms. As shown in Scheme 5A, according to the NCL reaction, the initial transthioesterification reaction between 2 and Cys would result in intermediate M1, and the subsequent intramolecular S,N-acyl shift would produce intermediate M2. Because 2, M1, and M2 all belong to coumarin-hemicyanine dye with the absorption wavelength around 550 nm, the observed absorption peak at 405 nm should be attributed to the seven-membered cyclic 2-Cys, a typical coumarin dye, produced via the intramolecular cyclization of M2. This speculation was supported by a control compound S2, whose absorption maximum was also observed around 405 nm (Fig. S3, ESI†). However, for Hcy, in terms of the almost unchanged absorption spectra of 2, the transthioesterification reaction should dominate, resulting in thioester 2-Hcy (Scheme 5B). To explain why it is difficult for 2-Hcy to perform the subsequent S,N-acyl shift, we proposed an electrostatic attraction interaction between the negatively charged Hcy carboxylate anion and the positively charged benzothiazolium N atom, which may prevent the Hcy amino group close to the thioester group, thereby inhibiting the subsequent S,N-acyl shift. In fact, the electrostatic interaction is sensitive to distance in space, and maybe it is more efficient for Hcy than for Cys to direct its carboxylate anion close to the positively charged benzothiazolium N atom. Similarly, in case of GSH, thioester 2-GSH produced via the direct transthioesterification reaction (Scheme 5C) should be the only product based on the almost unchanged absorption spectra, because the subsequent S,N-acyl shift must go through a ten-membered macrocyclic transition state that is obviously unstable.29,31 In fact, this speculation was also supported by the almost unchanged absorption spectra of 2 upon being treated with NAC (Fig. 4D). Furthermore, we also performed the HRMS assays, in which the molecular ion peaks corresponding to 2-Cys, 2-Hcy, and 2-GSH were clearly observed (Fig. S4, ESI†).
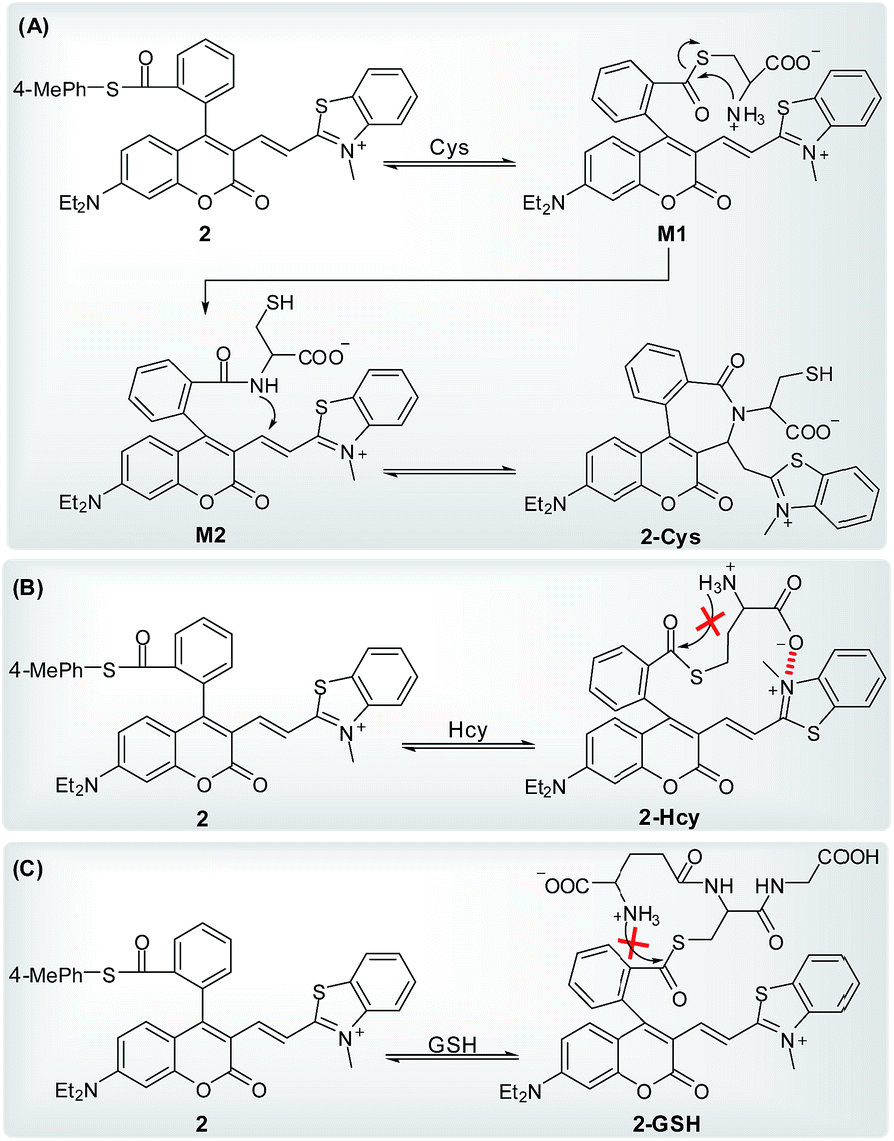 | ||
| Scheme 5 The proposed reaction mechanisms of 2 with Cys (A), Hcy (B), and GSH (C). | ||
In order to get insight into the above mentioned electrostatic interaction that inhibits 2-Hcy to undergo the subsequent S,N-acyl shift, DFT calculations with the B3LYP exchange functional employing 6-31G* basis sets using a suite of Gaussian 09 programs were conducted. As a comparison, the intermediate M1 resulting from Michael addition of Cys with 2 was also optimized. The optimized structures of 2-Hcy and M1 are shown in Fig. 5. As can be seen, in both the structures, the carboxylate anion is close to the positively charged benzothiazolium N atom, supporting our proposed electrostatic interaction. Notably, in the case of 2-Hcy (Fig. 5A), the distance between the carboxylate anion and benzothiazolium N atom is shorter than that in M1 (Fig. 5B), suggesting that its carboxylate anion could bind more tightly to the benzothiazolium N atom, thereby inhibiting the subsequent intramolecular S,N-acyl shift. However, in the case of M1, besides the relatively longer distance between the carboxylate anion and the benzothiazolium N atom, the coumarin-hemicyanine unit also showed a considerably twisted conformation, obviously different from the almost planar one found in 2-Hcy, revealing a poor electrostatic interaction, as well as an unstable molecular conformation. Possibly, in solution the electrostatic interaction in M1 would become weaker or disappear due to the strong salvation, as well as the tendency of restoring the planar and π-conjugated coumarin-hemicyanine conformation. Thus, it should be easier for M1 than for 2-Hcy to undergo the subsequent S,N-acyl shift. Although it is difficult for us to obtain some experimental evidences to support the proposed electrostatic interaction in 2-Hcy, the theoretical calculations perfectly rationalize our proposed reaction mechanisms. In fact, in some reports, the electrostatic interaction has been indicated to play a role in selectivity.23e,28b,c
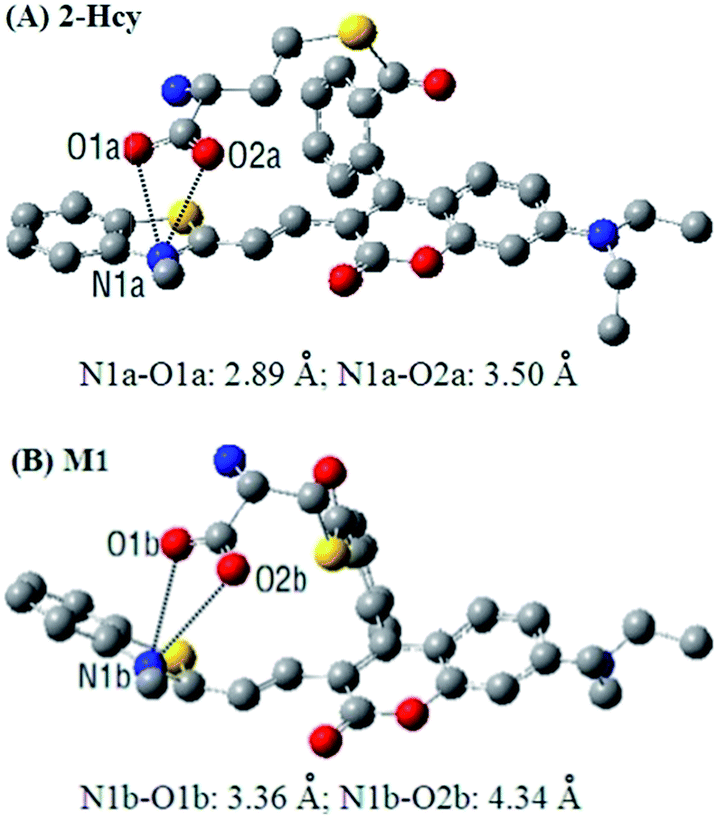 | ||
| Fig. 5 DFT optimized structures of 2-Hcy (A) and M1 (B) indicate the electrostatic interaction between the negatively charged carboxylate anion and the positively charged benzothiazolium N atom. In the ball-and-stick representation, carbon, nitrogen, and oxygen atoms are colored in grey, blue, and red, respectively. H atoms are omitted for clarity. | ||
In addition, it has been indicated that both Cys and Hcy can participate in a NCL reaction, but Cys appears to be slightly more efficient than Hcy.31 This can be attributed to the more stable five-membered cyclic transition state for Cys compared to the six-membered one for Hcy during the S,N-acyl shift in the NCL reaction. Probably, this is an additional contributing factor for the selectivity of probe 2 toward Cys over Hcy. However, efforts to identify 2-Cys adduct were unsuccessful due to the difficult separation. However, the UV-vis spectra changes of 2 treated with Cys (Fig. 4A), combined with the absorption spectrum of a control compound S2 (Fig. S3, ESI†) and the HRMS result of 2-Cys (Fig. S4, ESI†), strongly indicate the cyclic structure of 2-Cys proposed in Scheme 5A.
Encouraged by the above results, we examined emission behaviour of 2 upon the addition of Cys, Hcy, GSH, and NAC, respectively, by using two different excitations at 405 nm and 545 nm, equal to and near the absorption maximum of 2-Cys and 2, respectively. As shown in Fig. 6, the free probe displayed a poor emission at 494 nm (λex = 405 nm) (Fig. 6A1) and a strong emission at 644 nm (λex = 545 nm) (Fig. 6A2), suggesting that the probe exists as a ring-open form; upon the addition of 20 equiv. of Cys, the emission intensity at 494 nm gradually increased with the simultaneous decrease in the emission at 644 nm. After 30 min, the reaction reached equilibrium and, in this case, an approximate 96-fold enhancement of the ratiometric value (I494/I644) was observed. However, the addition of Hcy (or GSH) induced only a slight change in the ratiometric value [1.6 and 0.78-fold for Hcy (Fig. 6B1 and B2) and GSH (Fig. 6C1 and C2), respectively]. These results are in good agreement with the aforementioned absorption spectra changes, not only supporting our proposed reaction mechanisms, but also confirming the high selectivity of 2 for Cys over Hcy and GSH.
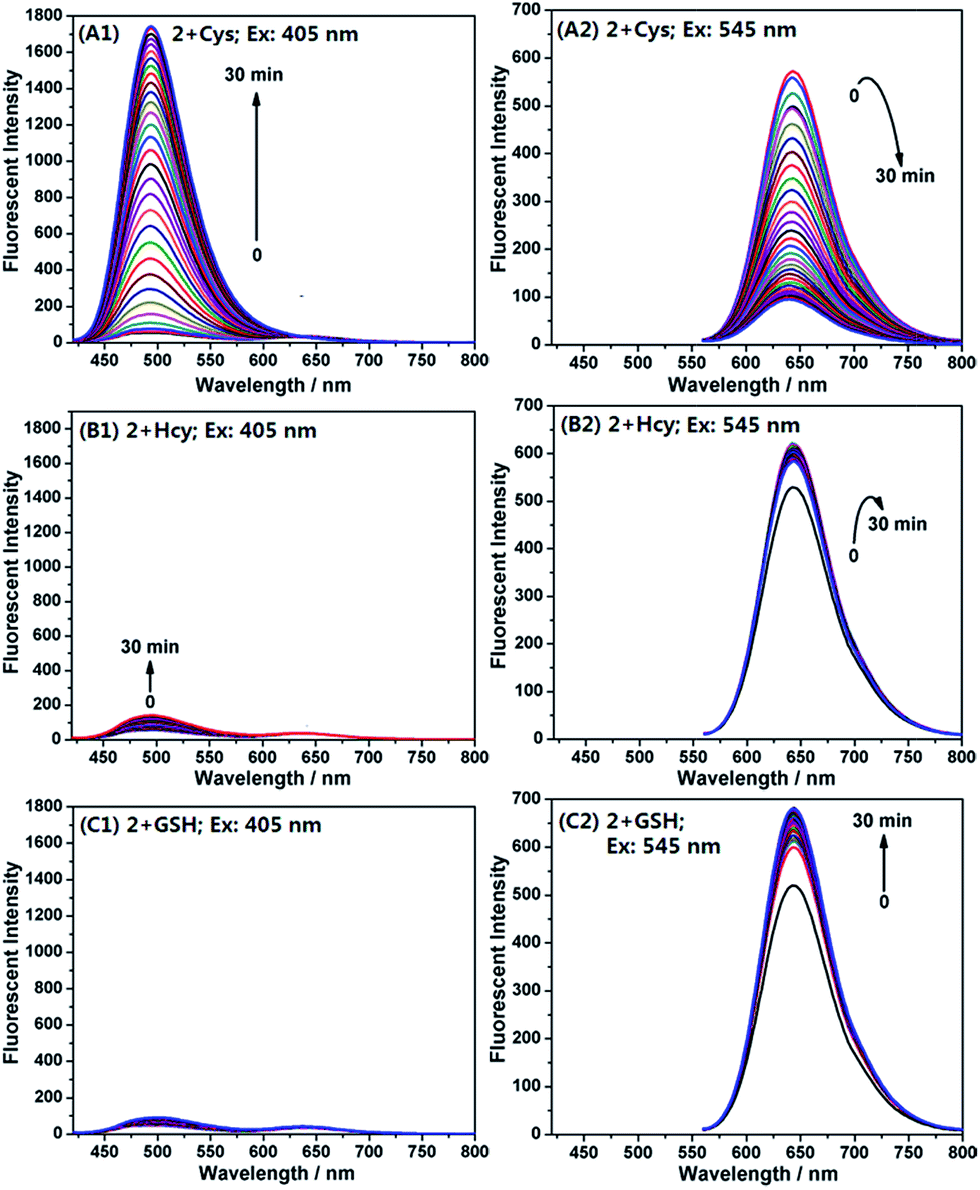 | ||
| Fig. 6 Time dependent fluorescence spectra of probe 2 (4 μM) in the presence of 20 equiv. of Cys (A1 and A2), Hcy (B1 and B2), and GSH (C1 and C2) in PB (pH 7.4, 20 mM, containing 10% DMF) at 37 °C. For A1–C1, λex = 405 nm, λem = 494 nm. For A2–C2, λex = 545 nm, λem = 644 nm. Slit: 5/10 nm. Voltage: 600 V. | ||
Furthermore, we speculated that the Cys-induced fluorescence response should also be able to be observed even in the presence of Hcy and GSH because the transthioesterification reaction between 2 and Hcy (or GSH) is reversible, but the intramolecular S,N-acyl shift reaction is irreversible (Scheme 5).14 As shown in Fig. S5 (ESI†), when the probe was treated with the mixture of Cys/Hcy (for each, 20 equiv.), we observed almost the same response as that treated with Cys only; a similar case was also observed when the mixture of Cys/GSH was used; moreover, when probe 2 was pre-treated with 20 equiv. of NAC for 30 min, and then treated with 20 equiv. of Cys, the obvious fluorescence responses toward Cys could also be observed. Because Cys, Hcy and GSH commonly coexist in biological systems, the present results are considerably attractive.
In addition, we also examined the fluorescence responses of 2 incubated with 20 equiv. of various amino acids, including His, Glu, Asp, Val, Phe, Tyr, Ala, Ser, Leu, Arg, Pro, Thr, Gln, Try, Ile, Lys, Cys, Hcy, and GSH. As can be seen in Fig. S6 (ESI†), Cys elicited significant fluorescence intensity changes at both 494 nm and 644 nm, whereas other amino acids caused only very limited emission changes or none at all in the same conditions. Thus, the selectivity of 2 toward Cys over Hcy, GSH, and other amino acids is considerably high. Moreover, when the solution of 2 was excited at 365 nm by UV lamp in the presence of 20 equiv. of various amino acids, only Cys caused a bright blue fluorescence discernible by naked eyes (Fig. 7).
 | ||
| Fig. 7 An image showing the emission of 2 (4 μM) treated with 20 equiv. of various amino acids in PB (pH 7.4, 20 mM, containing 10% DMF) at 37 °C. (1) 2 only, (2) His, (3) Glu, (4) Asp, (5) Val, (6) Phe, (7) Tyr, (8) Ala, (9) Ser, (10) Leu, (11) Arg, (12) Pro, (13) Thr, (14) Gln, (15) Try, (16) Ile, (17) Lys, (18) Cys, (19) Hcy, and (20) GSH. Ex = 365 nm. | ||
The fluorescence titration experiments for Cys were performed in the same condition. Upon treatment with the increasing concentrations of Cys, the fluorescence intensity of 2 at 494 nm gradually increased along with the simultaneous decrease of that at 644 nm, and when the amount of Cys was more than 80 μM, spectra saturation was reached. In this case, a linear calibration graph of the ratiometric responses (I494/I644) to Cys concentrations from 0 μM to 25 μM could be obtained (Fig. S7, ESI†), and the detection limit was measured to be 0.06 μM based on S/N = 3. Because the intracellular Cys concentrations are in the range of 30–200 μM,32 probe 2 is sensitive enough to image the biothiol in cells.
The effect of pH on the fluorescence response of probe 2 to Cys was also tested with the excitation wavelengths of 405 nm and 545 nm, respectively. It was found that 2 was stable over a wide pH range of 2–8 (Fig. S8A, ESI†), and displayed the obvious response for Cys in the region of 7–8 (Fig. S8B, ESI†). Thus, probe 2 could properly function at physiological pH.
Imaging Cys by 2 in living cells
Encouraged by the above in vitro assays, we subsequently evaluated the capability of probe 2 to image Cys in human kidney carcinoma cell line 786-O (Fig. 8). 786-O cells were found to have almost no fluorescence in blue channel. However, when 786-O cells were incubated with 2 (4 μM), they gave fluorescence in the channel (Fig. 8A1), suggesting that 2 is responsive to intracellular Cys. When 786-O cells were pre-treated with 0.5 mM Cys, and then incubated with 4 μM 2, an obvious emission enhancement in blue emission was observed (Fig. 8B1). When 786-O cells were pretreated with N-ethylmaleimide (NEM, 0.5 mM, a trapping reagent of thiol species), and then incubated with 2, a remarkable emission decrease in blue channel was observed (Fig. 8C1). These results suggested that probe 2 was cell permeable, and could selectively image Cys in living cells. In addition, the minimal cytotoxicity of 2 was also demonstrated by MTT assay (Fig. S9, ESI†).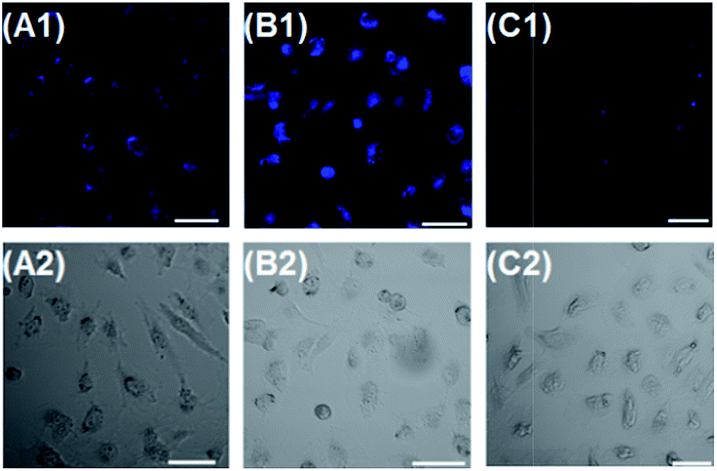 | ||
| Fig. 8 Imaging Cys in human kidney carcinoma cell line 786-O using probe 2. (A1) Incubated with 4 μM 2 only. (B1) Pre-treated with 0.5 mM Cys, and then 4 μM 2. (C1) Pre-treated with 0.5 mM NEM, and then 4 μM 2. (A2–C2) The corresponding bright field images. Emission was collected at 425–475 nm for blue channel (Ex: 405 nm). Scale bar: 50 μm. | ||
Conclusions
In this paper we presented a new carboxylic acid-functionalized coumarin-hemicyanine dye 1. The dye possesses good water solubility and attractive photophysical properties including red emission, good fluorescence quantum yield, large Stokes shift, and good photostability, and has been proved to be a suitable imaging agent for targeting mitochondria. With the dye platform, we further developed a ratiometric fluorescent probe 2, a thioester of 1, which can highly selectively detect Cys over Hcy and GSH based on the Cys-induced NCL-cyclization cascade reaction. Even in the presence of Hcy or GSH, 2 still works well with Cys due to the reversible transthioesterification reaction and the irreversible NCL reaction. The cell imaging experiment revealed that 2 is cell permeable, and could selectively image Cys in living cells. We expect that dye 1 or its counterparts will serve as a useful platform for the development of various fluorescent probes based on the cyclization or ring-open fluorescence switching mechanism. We also expect that the combination of the NCL-cyclization reaction with electrostatic interaction could be regarded as a useful design strategy to construct new fluorescent probes for biothiols with improved performance.Acknowledgements
We acknowledge the Natural Science Foundation of China (no. 21172137 and 21302114), Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP, 20101401110010), and Program for New Century Excellent Talents in University (NCET-11-1034) for support of this work.Notes and references
-
(a) G. Grynkiewicz, M. Poenie and R. Y. Tsien, J. Biol. Chem., 1985, 260, 3440 CAS
; (b) A. Minta, J. P. Y. Kao and R. Y. Tsien, J. Biol. Chem., 1989, 264, 8171 CAS
-
(a) E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517 CrossRef CAS PubMed
-
(a) Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS PubMed
- K. Kiyose, S. Aizawa, E. Sasaki, H. Kojima, K. Hanaoka, T. Terai, Y. Urano and T. Nagano, Chem.–Eur. J., 2009, 15, 9191 CrossRef CAS PubMed
-
(a) W. Shi and H. Ma, Chem. Commun., 2012, 48, 8732 RSC
-
(a) V. Dujols, F. Ford and A. W. Czarnik, J. Am. Chem. Soc., 1997, 119, 7386 CrossRef CAS
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316 CrossRef CAS PubMed
- Y.-Q. Sun, J. Liu, X. Lv, Y. Liu, Y. Zhao and W. Guo, Angew. Chem., Int. Ed., 2012, 51, 7634 CrossRef CAS PubMed
-
(a) Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, ACS Chem. Biol., 2011, 6, 600 CrossRef CAS PubMed
- T. Wang, Q.-J. Zhao, H.-G. Hu, S.-C. Yu, X. Liu, L. Liu, L. Liu and Q.-Y. Wu, Chem. Commun., 2012, 48, 8781 RSC
- L. Yuan, W. Lin, Y. Yang and H. Chen, J. Am. Chem. Soc., 2011, 134, 1200 CrossRef PubMed
- J. Chen, W. Liu, B. Zhou, G. Niu, H. Zhang, J. Wu, Y. Wang, W. Ju and P. Wang, J. Org. Chem., 2013, 78, 6121 CrossRef CAS PubMed
- I. Johnson and M. T. Z. Spence, The Molecular Probes Handbook
![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/char_e0d5.gif) A Guide to Fluorescent Probes and Labeling Technologies, LifeTechnologies, Carlsbad, CA, 11th edn, 2010 Search PubMed
A Guide to Fluorescent Probes and Labeling Technologies, LifeTechnologies, Carlsbad, CA, 11th edn, 2010 Search PubMed -
(a) P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776 CAS
-
(a) K. G. Reddie and K. S. Carroll, Curr. Opin. Chem. Biol., 2008, 12, 746 CrossRef CAS PubMed
-
(a) H. Bradley, A. Gough, R. S. Sokhi, A. Hassell, R. Waring and P. J. Emery, Rheumatology, 1994, 21, 1192 CAS
- M. Kondo, M. Tanaka, N. Sakamoto and H. Ooyoshi, Eur. Pat. EP511, 019, Mitsui Petrochemical Industries Ltd., 1993
-
(a) H. M. McBridel, M. Neuspiel and S. Wasiak, Curr. Biol., 2006, 16, 551 CrossRef PubMed
-
(a) M. Poot, Y. Zhang, J. A. Krämer, K. S. Wells, L. J. Jones, D. K. Hanzel, A. G. Lugade, V. L. Singer and R. P. Haugland, J. Histochem. Cytochem., 1996, 44, 1363 CrossRef CAS PubMed
-
(a) A. T. Hoye, J. E. Davoren, P. Wipf, M. P. Fink and V. E. Kagan, Acc. Chem. Res., 2008, 41, 87 CrossRef CAS PubMed
-
(a) X. Chen, Y. Zhou, X. Peng and J. Yoon, Chem. Soc. Rev., 2010, 39, 2120 RSC
-
(a) H. Chen, Q. Zhao, Y. Wu, F. Li, H. Yang, T. Yi and C. Huang, Inorg. Chem., 2007, 46, 11075 CrossRef CAS PubMed
-
(a) N. Shao, J. Jin, H. Wang, J. Zheng, R. Yang, W. Chan and Z. Abliz, J. Am. Chem. Soc., 2010, 132, 725 CrossRef CAS PubMed
-
(a) O. Rusin, N. N. St Luce, R. A. Agbaria, J. O. Escobedo, S. Jiang, I. M. Warner, F. B. Dawan, K. Lian and R. M. Strongin, J. Am. Chem. Soc., 2004, 126, 438 CrossRef CAS PubMed
-
(a) F. Kong, R. Liu, R. Chu, X. Wang, K. Xu and B. Tang, Chem. Commun., 2013, 49, 9176 RSC
-
(a) H. Li, J. Fan, J. Wang, M. Tian, J. Du, S. Sun, P. Sun and X. Peng, Chem. Commun., 2009, 39, 5904 RSC
-
(a) Z. Guo, S. W. Nam, S. Park and J. Yoon, Chem. Sci., 2012, 3, 2760 RSC
-
(a) H. S. Jung, J. H. Han, T. Pradhan, S. Kim, S. W. Lee, J. L. Sessler, T. W. Kim, C. Kang and J. S. Kim, Biomaterials, 2012, 33, 945 CrossRef CAS PubMed
-
(a) L.-Y. Niu, Y.-S. Guan, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Commun., 2013, 49, 1294 RSC
-
(a) L. Long, W. Lin, B. Chen, W. Gao and L. Yuan, Chem. Commun., 2011, 47, 893 RSC
-
(a) X.-F. Yang, Q. Huang, Y. Zhong, Z. Li, H. Li, M. Lowry, J. O. Escobedo and R. M. Strongin, Chem. Sci., 2014, 5, 2177 RSC
-
(a) T. K. Chung, M. A. Funk and D. H. Baker, J. Nutr., 1990, 120, 158 CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplemental spectra, and the 1H-, 13C-NMR, and MS spectrum. See DOI: 10.1039/c4ra10865e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |