
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Applying thermo-destabilization of microemulsions as a new method for co-catalyst loading on mesoporous polymeric carbon nitride – towards large scale applications†
M.
Schröder
,
K.
Kailasam
,
S.
Rudi
,
K.
Fündling
,
J.
Rieß
,
M.
Lublow
,
A.
Thomas
,
R.
Schomäcker
and
M.
Schwarze
*
Technische Universität Berlin, Institut für Chemie, Straße des 17. Juni 124, 10623 Berlin, Germany. E-mail: ms@chem.tu-berlin.de
First published on 7th October 2014
Abstract
Photocatalytic water reduction using polymeric carbon nitride is an emerging research area, but other issues like the co-catalyst loading and the choice of sacrificial agents as hole scavengers have received very little attention in the literature. We addressed the above issues by employing microemulsion based ex situ co-catalyst loading on porous carbon nitrides as a superior method with about 40% higher HER activity compared to the usual in situ co-catalyst loading used in the majority of previous studies. In addition, for the first time, we introduced a large scale 1 m2 reactor for H2 production with 80 mL h−1 using this ex situ Pt loaded on porous carbon nitrides under natural sunlight conditions.
1. Introduction
Due to their enormous potential as heterogeneous, metal free photocatalysts and their capability for water reduction, polymeric carbon nitrides, CN (also commonly called “g-C3N4”) have been the subject of tremendous research activity over recent years.1–4Carbon nitrides, which are prepared by pyrolysis of nitrogen-rich precursors such as cyanamide, dicyandiamide or melamine, have a low surface area (∼10 m2 g−1) when prepared without any template. This bulk carbon nitride shows a relatively low activity for photocatalytic hydrogen evolution in the presence of sacrificial oxidant.3 Numerous attempts have been made to improve the performance of carbon nitrides, for example introducing porosity and higher surface areas by hard templating which indeed largely enhanced the photocatalytic activity.5–9 Several other routes like chemical modification using various organic molecules by copolymerization, blending of conjugated moieties or by macroscopic assembly of the precursors also increase their performance for water reduction.10–18
It has been recently reported that mesoporous carbon nitrides (mp-CN) can be prepared by a one-pot synthesis using a combined sol–gel/thermal condensation method, which showed high activity for H2 evolution in the presence of a sacrificial oxidant.12,19–22 These materials are superior in activity when compared to other porous carbon nitrides prepared by two-step hard templating approaches with silica nanoparticles or anodic alumina membranes.7–9,23,24
However, beside all these structural variations on CNs the addition of a suitable co-catalyst is essential to attain high values for hydrogen evolution from water, in order to reduce the overpotential for the reduction reaction.3 Indeed, with the carbon nitride alone just very low amounts of H2 can be evolved from water.3 Platinum is used as co-catalyst in most studies, while in some cases Ni, Co, Cu, Au, and Ag based catalysts are used to enhance the activity for H2 evolution.25–30 However, Pt co-catalysts are superior to any other metal ions/hydroxides/oxides in the water reduction reaction.
Usually Pt and other co-catalysts are deposited on the carbon nitride structure by an in situ photodeposition method, in which the platinum precursor is simply added in certain amounts (mostly about 3 wt% Pt) to the reaction solution. Pt nanoparticles (Pt NPs) are then deposited on the surface of the CN materials by photoreduction. Mostly the Pt precursor is added in excess, which results in obscuring of the reaction solution by the formation of dispersed Pt nanoparticles (see Fig. S1†). Therefore, the photocatalyst absorbs less light, causing decrease in activity in the long run. So far no studies have looked into this issue (except by employing non-ionic Pt precursor),31 even though examination of the optimum Pt content is highly important, especially to transfer this process to large scale applications with low cost photocatalysts like carbon nitrides. Thus, while a certain amount of Pt is needed to increase the photocatalytic activity of the overall system, this is of course accompanied by increasing costs and, as stated above, when a certain value is reached can even be detrimental to the activity. Beside the pure Pt amount, another important point is that small nanoparticle sizes and a good dispersion of the co-catalyst on the photocatalyst surface have to be ensured to achieve enhanced activity. None of these issues have been described in detail up to now but certainly need to be considered, as they define the number of “active catalytic centers” of the overall catalyst.
One way to make better use of the Pt and to avoid large amounts of unsupported, inactive Pt NPs in solution is to use an ex situ process. Photodeposition in certain solvents and wet-impregnation are ex situ methods often used for the preparation of supported catalysts, but the dispersion of the nanoparticles is often inhomogeneous and combined with a high degree of particle agglomeration. Therefore, a microemulsion technique followed by photoreduction to achieve uniform Pt dispersion with small Pt NPs on the surface of porous CN materials was used instead. Microemulsions are excellent media for the synthesis of metal nanoparticles and allow for size and shape control of Pt NPs.32 The first formed NPs are then impregnated onto the support directly from the microemulsion via thermo-destabilization, a new method which was recently developed for the deposition of Pt nanoparticles on alumina surfaces.33,34 In this report, we employed this technique to prepare Pt NPs and also other noble metal NPs such as Rh and Pd on the porous carbon nitride networks. For comparison also the standard in situ photodeposition method was used to prepare the Pt co-catalyst. Most of the earlier reports on photocatalysis using CN materials use high power Xe-lamps as light sources, but for future commercial applications it is crucial that real sunlight can be used. In this report, we show that photocatalytic activity can be observed using natural sunlight as light source.
Another important task for future applications is to transform the lab-scale photocatalytic process to a pilot scale process. In this work, a large-scale dispersion setup using porous carbon nitrides for large scale H2 production in the presence of natural sunlight is described which can pave the way for possible future technologies to target growing energy demands.
2. Experimental part
2.1. Chemicals for thermo-destabilization of microemulsions
In the thermo-destabilization of microemulsions the following chemicals were used without further purification: cyclohexane (≥99.5%, Carl-Roth) as the oil phase, distilled water, Triton X-100 (100%, Sigma-Aldrich) as the surfactant, 1-pentanol (≥98%, Carl-Roth) as the co-surfactant, chloroplatinic acid solution (8 wt% in H2O, Sigma-Aldrich) as precursor for platinum nanoparticles as co-catalyst, ascorbic acid (>99%, Alfa Aesar) as the reducing agent and acetone (≥99.8%, Carl Roth) as the washing agent. Mesoporous carbon nitride (mp-CN) (see paragraph 2.3) was used as Photocatalyst.2.2. Chemicals for photocatalytic water reduction
For photocatalytic water reduction experiments, triethanolamine (TEOA, ≥99% purity, Sigma-Aldrich) was used as sacrificial agent and distilled water was used as hydrogen source. All chemicals were used without any further purification.2.3. Thermo-destabilization of microemulsions
| α = moil/(moil + mwater) |
| γ = (msurfactant + mco-surfactant)/(moil + mwater + msurfactant + mco-surfactant) |
| δ = mco-surfactant/(msurfactant + mco-surfactant) |
In our standard experiment, α, γ and δ were equal to 0.75, 0.4 and 0.5, respectively.
For the nanoparticle formation, the ascorbic acid containing microemulsion was added by a micropump (0.2 mL s−1) to the glass reactor, which already contained the microemulsion with the metal precursor. The reaction mixture was stirred at 720 rpm for 1 h at 298 K to form the nanoparticles. Subsequently, 200 mg carbon nitride was added and the microemulsion was heated up to 318 K and stirred for 2 h. During this time, the microemulsion turned into a two-phase system, releasing the nanoparticles to the carbon nitride. It must be mentioned that for Pt no nanoparticles were deposited on the carbon nitride surface due to the negative zeta potentials of both the carbon nitride and the Pt NPs as described in more detail in Section 3.2. For Pt as co-catalyst, only the precursor was adsorbed on the carbon nitride. After the deposition process or rather for Pt the adsorption process, the reactor was cooled down to 298 K, the co-catalyst loaded carbon nitride was separated from the reaction mixture by centrifugation at 8500 rpm for 15 min, the catalyst was washed three times with acetone and dried under vacuum at 353 K for 24 h. A more detailed description is given in the publication of Parapat et al.33
2.4. Hydrogen evolution experiments
In this investigation the initial co-catalyst concentration was varied in a range from 0 to 5 wt% Pt (0–11 wt% H2PtCl6) and the TEOA concentration was varied in a range from 2.5 to 60 vol%.
2.5. Analysis of photocatalyst, reaction mixture and evolved gases
Porous CN powders were fixed to XPS sample holders by pressing them onto adhesive and UHV-stable carbon tape. XPS was carried out at pass energies of 50 eV to increase the detection sensitivity for the minute Pt concentrations. Thereby, broadening of the energy dissipative curves occurred. A Mg K(alpha) X-ray source (1253.6 eV) was used for probing the chemical state of the surfaces. The X-ray instability of the powders was mitigated by reducing the power of the X-ray source down to 100 W (compared to typical power values of 250 W). Evaluation of the oxygen Auger line (KL23–KL23) at 745 eV suggests charging of the powders during XPS analysis, resulting in an apparent shift of the binding energies by about 0.5–0.8 eV. An Agilent 7890A gas chromatograph was used to determine the hydrogen content in the headspace of the reactor. The GC was equipped with a Carboxen-1000 column and a thermal conductivity detector (TCD). The carrier gas was argon (30 mL min−1). The contents of platinum on the catalysts were measured by inductive coupled plasma emission spectroscopy (ICP-OES) using an ICP-OES 517 (Varian Inc., USA). Prior to ICP-OES analysis the photocatalysts were pretreated by microwave (Discover SP-D, CEM, USA) digestion to dissolve the platinum nanoparticles. For calibration of the set-up, commercial standard solutions of platinum (1000 mg L−1, Aldrich) were diluted with distilled water to obtain concentrations of 2, 4.5, and 7.5 mg L−1. Platinum was analyzed at 203.464, 214.424, 224.552 and 265.945 nm. Transmission Electron Microscopy (TEM) measurements were operated by a FEI Tecnai G2 20 S-TWIN transmission electron microscope equipped with a LaB6-source at 200 kV acceleration voltages. For TEM-investigations a small amount of the sample powder was placed on a TEM-grid (carbon film on 300 mesh Cu-grid, Plano GmbH, Germany) and was sputtered with carbon. Nitrogen sorption analyses were carried out at the temperature of liquid nitrogen on a Quadrasorb instrument (Quantachrome) after evacuating the samples at 150 °C overnight. The surface areas and pore size distribution analysis were Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively. The pore volume was calculated at the relative pressure 0.99. For UV/Vis measurements a Lambda 35 UV/Vis spectrometer from Perkin Elmer (USA) was used. The surface morphology of the mp-CN was analyzed by scanning electron microscope (JEOL 7401F) at an acceleration voltage of 10 kV and at a working distance of 9 mm.3. Results and discussion
The photocatalytic activity of semiconducting materials strongly depends on the properties of the photocatalyst and on the amount, distribution and properties of the loaded co-catalyst. For photocatalytic hydrogen evolution in most cases, the co-catalyst is loaded by in situ photodeposition. However, it has been shown for mesoporous carbon nitrides (mp-CN) that the in situ photoreduction is strongly affected by the chosen reaction conditions and yields agglomerated nanoparticles and inhomogeneous nanoparticle distributions.22 Therefore, ex situ methods like wet impregnation35 or chemical reduction of metal precursors36 seems to be more appropriate for co-catalyst deposition. Here, we investigated a new ex situ approach based on microemulsion derived Pt NPs for the synthesis of highly distributed and well defined nanoparticles, functioning as co-catalyst on mp-CN.3.1. Synthesis and characterization of mp-CN photocatalysts
Mesoporous graphitic carbon nitride, mp-CN, was prepared using a published procedure.7 In short, mp-CN was prepared by a sol–gel/thermal condensation approach, where the carbon nitride precursor cyanamide (CA) was added to a HCl–ethanol solution. After dissolving the cyanamide, the pH was adjusted to 2 and tetraethylorthosilicate (TEOS) was added, and stirred for one hour (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 molar ratio of TEOS:CA). After the removal of the solvents, the composite gel was aged at 80 °C, thermally treated at 550 °C to obtain a carbon nitride-silica composite. Finally carbon nitride, mp-CN, is obtained by the removal of silica using NH4HF2 solution.
:6 molar ratio of TEOS:CA). After the removal of the solvents, the composite gel was aged at 80 °C, thermally treated at 550 °C to obtain a carbon nitride-silica composite. Finally carbon nitride, mp-CN, is obtained by the removal of silica using NH4HF2 solution.
The porous carbon nitride mp-CN was characterized using different techniques, showing the same features as reported before,7i.e. a peak at 27.4° in the powder XRD (Fig. S2a†) confirming the layered structure of the material and a broad absorption starting at 450 nm in the UV/Vis spectrum (Fig. 2Sb†). Nitrogen sorption measurements show type IV isotherm typical for mesoporous materials with surface area of about 149 m2 g−1 (Fig. S2c†), pore diameter of 38 Å (Fig. S1d†) and total pore volume of 0.28 cm3 g−1.
3.2. Synthesis and characterization of co-catalyst loaded carbon nitride
In recent years it has been shown that the Schottky effect plays an important role in capturing excited electrons on the surface of photocatalytic active materials.22,37 Therefore, the deposition of a defined amount of co-catalyst on the semiconductors surface can improve the photocatalytic activity significantly. For photocatalytic hydrogen evolution with carbon nitrides, several precious metals have been used as co-catalyst which clearly improved the photocatalytic performance.4 However, until now platinum shows the highest activity in photocatalytic hydrogen evolution applying carbon nitride materials as catalyst. It is expected that the hydrogen elimination from Pt–H surface bonds is favored in comparison to the covalent elimination from the CN surface.3 For platinum deposition, in most of the previous studies the in situ photoreduction is used. Therein, a metal precursor, usually hexachloroplatinic acid (H2PtCl6), is mixed with the photocatalyst in the reaction solution and due to irradiation the platinum ions get photochemically reduced and deposited on the carbon nitride surface. The advantages of this method are the easy experimental feasibility and that no further preparations are required. However, the in situ photoreduction for Pt loaded carbon nitride results in inhomogeneous distribution and highly agglomerated nanoparticles on the CN surface (Fig. 1a), with an average particle size between 10 to 30 nm. Due to the broad size distribution and high agglomeration it can be expected that the number of active sites on the surface of the photocatalyst and thereby the photocatalytic activity is reduced. Moreover, the inhomogeneous catalyst dispersion affects the photocatalytic activity to a large extent. Apart from limitations in photocatalytic activity, the difficult realization of a scale-up is another disadvantage. A more detailed description of the influences of the in situ photoreduction on the photocatalytic activity with carbon nitride materials was recently described by Schröder et al.22 | ||
| Fig. 1 TEM images of (a) Pt@mp-CN (1.2 wt% Pt) for in situ photodeposition, (b) Pd@mp-CN (1.1 wt% Pd) after ex situ microemulsion approach, (c) Pt@mp-CN (1.4 wt% Pt) after ex situ microemulsion approach, (d) after photocatalytic reaction and (e) after calcination at 573 K. | ||
Therefore, to obtain well defined and highly distributed platinum nanoparticles on mp-CN photocatalysts with the possibility for application on a large scale, a new ex situ microemulsion method was applied. This method was first reported by Parapat et al. for the preparation of supported Pt catalysts (on α-Al2O3, SiO2 and SBA-15) but has so far not been reported for applications in photocatalysis.38 For nanoparticle deposition by this method, first a metal precursor is reduced by a reduction agent in a reverse micellar system and then the formed nanoparticles are deposited onto the support material, here mesoporous carbon nitrides (mp-CN), after heating the solution above the phase transition temperature (for details see the Experimental part). In the beginning of our investigations we applied this method only for platinum as co-catalyst, with H2PtCl6 as metal precursor and ascorbic acid as reducing agent. After the first synthesis, the catalyst was analyzed using TEM and the result is shown in Fig. 1c. As can be seen from the image, no nanoparticles were observed, while EDX-measurements (Fig. S3†) clearly indicates Pt located on the surface of the carbon nitride support. However, after calcination (Fig. 1e) or after photocatalytic reaction (Fig. 1d) Pt nanoparticles appear on the surface of the ex situ treated photocatalyst.
It should be noted that the zeta-potentials of the catalyst surfaces and metal nanoparticles were found to be crucial factors for the successful loading of the nanoparticles onto the support material.22 Both the Pt NPs and carbon nitride have negative zeta-potentials, resulting in repulsions during the deposition procedure. This results in a lower amount of Pt NPs observed on the surface than expected from the initial loading of Pt. In contrast, Pd nanoparticles have a positive zeta-potential resulting in higher amount of nanoparticles on the surface due to stronger attraction to the carbon nitride surface (shown in Fig. 1b).
To investigate the formation of nanoparticles on the CN surface, we analyzed the support material, after applying the ex situ microemulsion approach, in X-ray photoelectron spectroscopy (XPS). The result is shown in Fig. 2. After ex situ preparation, the XPS shows a double-peak at binding energy values of 74 and 78 eV, which corresponds to Pt(IV). No Pt(0) formation was observed and together with the signal for Cl (not shown), it was clarified that only adsorbed H2PtCl6 are deposited on the surface. After testing this catalyst in HER, Pt(0) signal was obtained in XPS (double peak at about 71.5 and 75 eV) due to photoreduction of the adsorbed precursor. It should be mentioned that a similar double-peak is observed for in situ co-catalyst loaded Pt. These results are supported by earlier studies on Pt@TiO2 which shows a double-peak at 70.9 and 74.3 eV.39
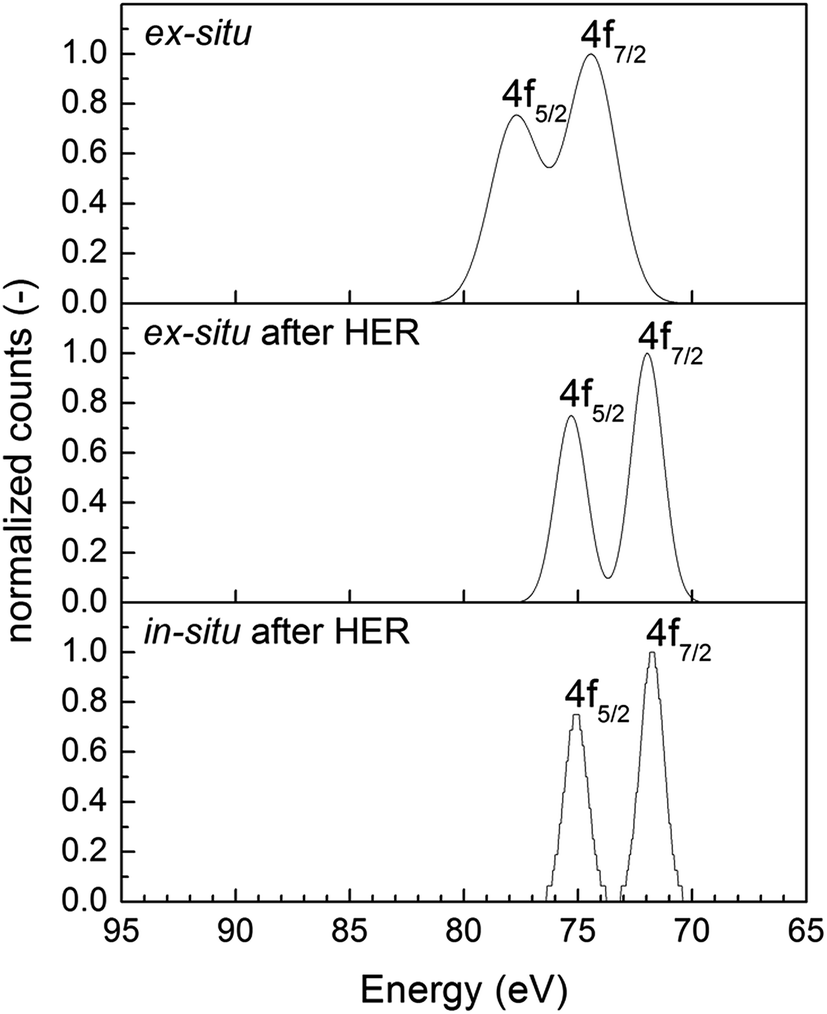 | ||
| Fig. 2 XPS of ex situ prepared Pt@mp-CN (top), after applying ex situ Pt@mp-CN in HER (middle) and in situ prepared Pt@mp-CN. | ||
After photocatalytic reaction highly dispersed nanoparticles with an average diameter smaller than 5 nm are obtained (see Fig. 1d) compared to the in situ loaded catalyst where larger and agglomerated nanoparticles are observed (Fig. 1a). This observation on the in situ and the ex situ loaded mp-CN was reflected from their photocatalytic activity. To compare the photocatalytic activity and the optimal Pt loading of the in situ and the ex situ treated mp-CN, the hydrogen evolution rate as function of the initial Pt concentration is shown in Fig. 3.
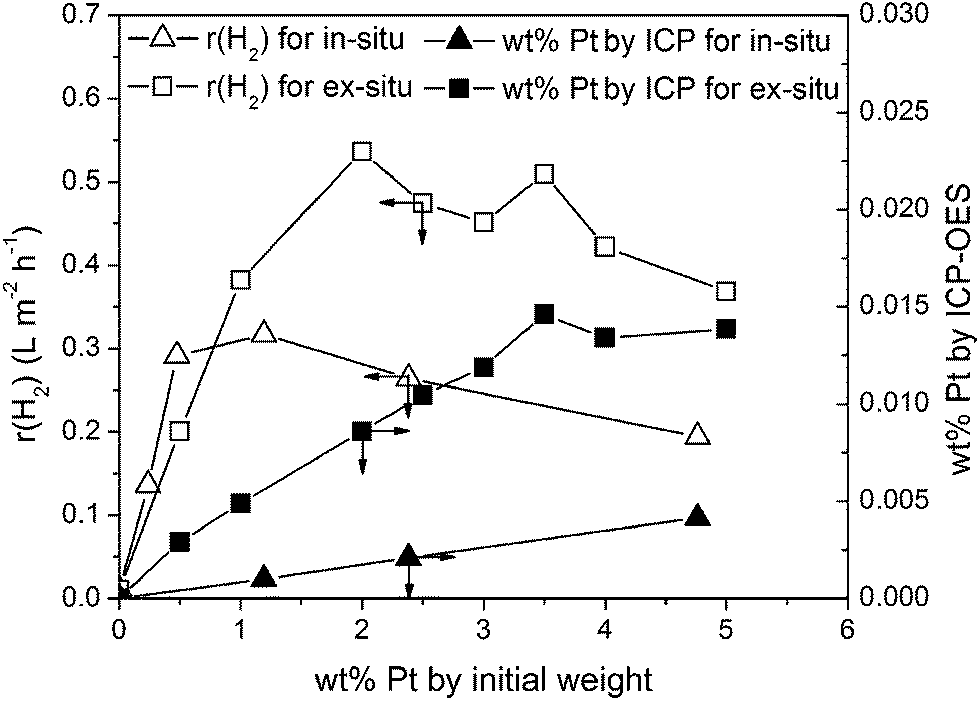 | ||
| Fig. 3 H2 evolution rate and Pt loading as a function of the initial Pt concentration for in situ and ex situ treated mp-CN (1.3 g L−1 mp-CN, 10 vol% TEOA, 298 K, solar simulator 1000 W m−2). | ||
For the ex situ treated carbon nitride the optimal initial platinum concentration is between 1.9 and 3.6 wt% and peak activity is reached at a hydrogen evolution rate of 0.53 L m−2 h−1. The photocatalytic activity of the in situ loaded mp-CN reaches peak activity at 0.32 L m−2 h−1. By comparing the activities of both approaches, Fig. 3 illustrates that the highest hydrogen evolution rate of the ex situ treated mp-CN is about 40% (±10%) higher. This led to the assumption that the better distribution of the nanoparticles by the microemulsion approach leads to increased photocatalytic activity. The increase in activity between ex situ and in situ was confirmed by using different carbon nitride batches.
The optimal initial Pt loading for achieving maximum photocatalytic activity of the in situ loaded mp-CN is at 1.2 wt%. However, it was shown before that the optimal initial weight fraction of Pt for in situ photoreduction also depends on the batch of the mesoporous carbon nitride and is in a range between 1 and 5 wt%.22 This is corroborated by studies of Wang et al. showing that the optimal initial Pt concentration for graphitic carbon nitride is between 2 and 4 wt%.3 Therefore, the ex situ pathway requires nearly the same initial Pt concentration. To obtain the effective Pt loading on the surface of the carbon nitride support, we analyzed the Pt loaded photocatalysts after photocatalytic reaction by ICP-OES. It is shown that the effective platinum loading strongly differs from the initial Pt amount, due to the similar negative zeta potentials for both carbon nitride and Pt NPs as mentioned above. Therefore, only low loading of Pt was obtained on the carbon nitride. However, Fig. 3 illustrates that for the same initial weight fraction, the effective Pt loading for the ex situ treated carbon nitride is about ten times higher. Probably, this effect is caused by the protective barrier of the micelles, which build an electrostatic shield and weaken the rejection between the nanoparticles and the CN material resulting in higher loading of nanoparticles. For comparison we prepared one catalyst using the impregnation method with 2 wt% Pt loading, but activity of this catalyst was about 40% (±10%) lower than the activity of the catalyst prepared via the microemulsion approach.
Beside photoreduction during photocatalytic reaction, calcination is an alternative pathway to reduce the adsorbed platinum precursor on the carbon nitride surface. After calcination, the nanoparticles are even smaller and better dispersed (see Fig. 1d), but the photocatalytic activity is reduced as shown in Table 1. This may be speculated to be caused by the formation of more condensed structure and changing properties of the carbon nitride semiconductor and further investigations are going on in our research to clarify this.
| Calcination temperature (K) | r(H2) (L m−2 h−1) |
|---|---|
| No calcinations | 0.410 |
| 423.15 | 0.227 |
| 573.15 | 0.082 |
| 723.15 | 0.009 |
After the optimization of the platinum concentration on mp-CN along with higher photocatalytic H2 evolution by ex situ method, the amount of photocatalyst in ex situ synthesis was increased to obtain higher amounts of Pt-loaded carbon nitride for large scale hydrogen production. Therefore, we varied the amount of photocatalyst from 200 to 2000 mg and measured the photocatalytic activity for HER. Table 2 illustrates that the photocatalytic activity varies only in a narrow range showing the superiority of this method when compared to the in situ method, where the loading of Pt cannot be controlled. Thus, this ex situ method seems to be an appropriate synthesis strategy for Pt loaded carbon nitride which could be applied in large scale hydrogen production. Below we present a new “large scale setup” for hydrogen evolution under real sunlight conditions and show the activity for the ex situ prepared mp-CN photocatalyst.
For further improvement of the HER with mesoporous carbon nitride, the microemulsion approach was applied to load nanoparticles of various metals as co-catalyst (Pd, Ru and Rh). It was found that although the ex situ microemulsion approach is transferable for loading different co-catalysts on the surface of a photocatalyst, the activity for HER is very low when compared to Pt as co-catalyst. It would be interesting to see how the loading of different metal precursors would affect the size, concentration and distribution of the metal nanoparticles, but this is beyond the scope of the present study.
3.3. Impact of the reaction conditions on the photocatalytic activity
Recently we have shown that the reaction conditions have a high impact on the in situ photoreduction and thereby on the photocatalytic activity.22 Therein, the triethanolamine concentration and the platinum concentration were shown to have the highest impact on the activity. The influence of the platinum concentration was already discussed before. Here, we want to investigate the influence of the TEOA concentration, where the alcohol acts as electron donor in the photocatalytic water reduction reaction.Fig. 4 shows that in the absence of TEOA, no H2 evolution is observed for the ex situ as well as for the in situ platinum loaded carbon nitride. With increasing TEOA concentration the hydrogen evolution rate increases strongly up to a maximum activity. For the in situ loaded carbon nitride the maximum rate is obtained for a TEOA concentration of 30 vol% and beyond this concentration the activity decreases substantially. This trend is due to the strong impact of the TEOA concentration on the in situ photoreduction process of H2PtCl6. With an increasing TEOA concentration, the effective platinum loading on the carbon nitride surface is reduced and thereby the optimal platinum loading falls below a certain limit.22 Therefore, the hydrogen evolution rate decreases substantially with increasing TEOA concentration after highest photocatalytic activity at 30 vol% TEOA concentration. For the ex situ platinum loaded mp-CN the hydrogen evolution rate stays constant and reaches a plateau beyond a concentration of 5 vol%. This trend for the photocatalytic activity is in accordance with the Langmuir–Hinshelwood type rate law and is similar to the trend observed by Li et al.40 Thus, it is shown that the impact of the higher sacrificial agent concentration could be overcome by applying the ex situ microemulsion approach.
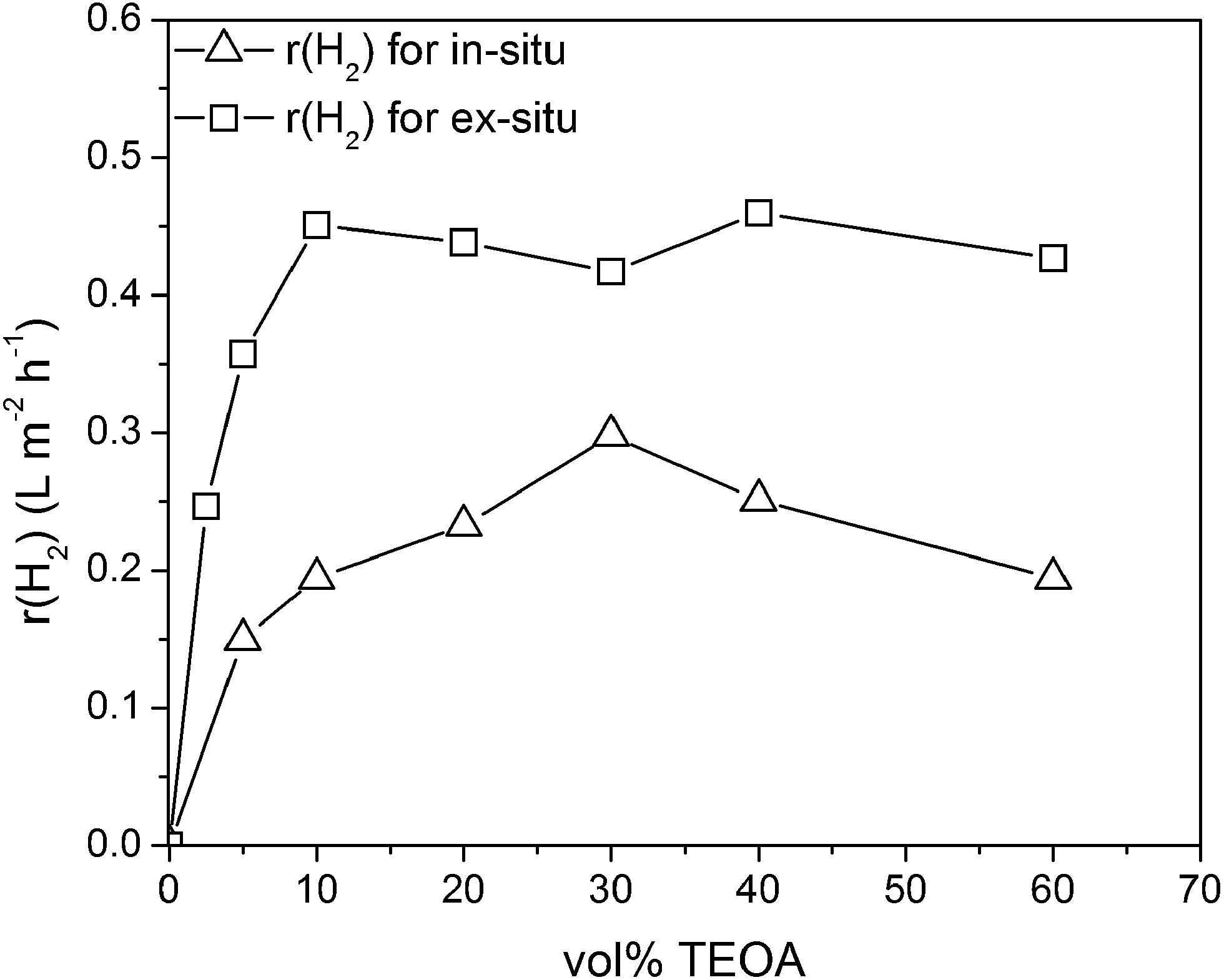 | ||
| Fig. 4 H2 evolution rate as a function of the initial TEOA-concentration for ex situ and in situ loaded mp-CN (1.3 g L−1 mp-CN, 2 wt% Pt for ex situ and 4.8 wt% Pt for in situ related to mp-CN, 298 K, solar simulator 1000 W m−2). | ||
Table 3 shows the photocatalytic activity of various sacrificial agents used in HER with the ex situ loaded carbon nitrides. The highest rates are obtained with triethanolamine followed by EDTA, which is probably due to strong H-bonding interaction of the hydroxyl and carboxyl groups of TEOA and EDTA with the nitrogen rich surface of the carbon nitride. For other alcohols the activity is strongly reduced. However, with the in situ Pt-loaded mp-CN, we obtained a significant hydrogen evolution rate only with TEOA.
| Entry | Sacrificial Agent (SA) | Normalized ratea |
|---|---|---|
| a Based on hydrogen evolution rate of carbon nitride with TEOA as SA. | ||
| 1 | Triethanolamine (TEOA) | 1.000 |
| 2 | Ethylenediaminetetraacetic acid (EDTA) | 0.830 |
| 3 | Methoxybenzaldehyde (MeOBA) | 0.140 |
| 4 | Benzaldehyde (BA) | 0.111 |
| 5 | Methanol (MeOH) | 0.028 |
| 6 | Ethanol (EtOH) | 0.007 |
| 7 | Butanol (BuOH) | 0.007 |
3.4. Large scale hydrogen production with ex situ prepared mp-CN
There are different types of photoreactors to perform sacrificial hydrogen evolution with light being injected from the top,41,42 from the side43 or even from inside.44 It is also possible to use a membrane reactor and perform Z-type overall water splitting with two photocatalysts and an electron mediator.45 However, all these reactors are used for lab scale experiments. Prior to our attempts to develop a large scale setup for hydrogen evolution, for comparison, we determined the activity of Pt@mp-CN in the lab-scale setup under optimized conditions (0.42 L h−1 m−2) and with a small demonstrator (Fig. S5†) it was proven that hydrogen evolution with real sunlight is possible (Fig. 5). Thereafter, based on our lab scale photoreactor with defined geometry, we developed a setup for large scale hydrogen evolution which is shown in Fig. 6, using the irradiation area as the main scale-up criterion.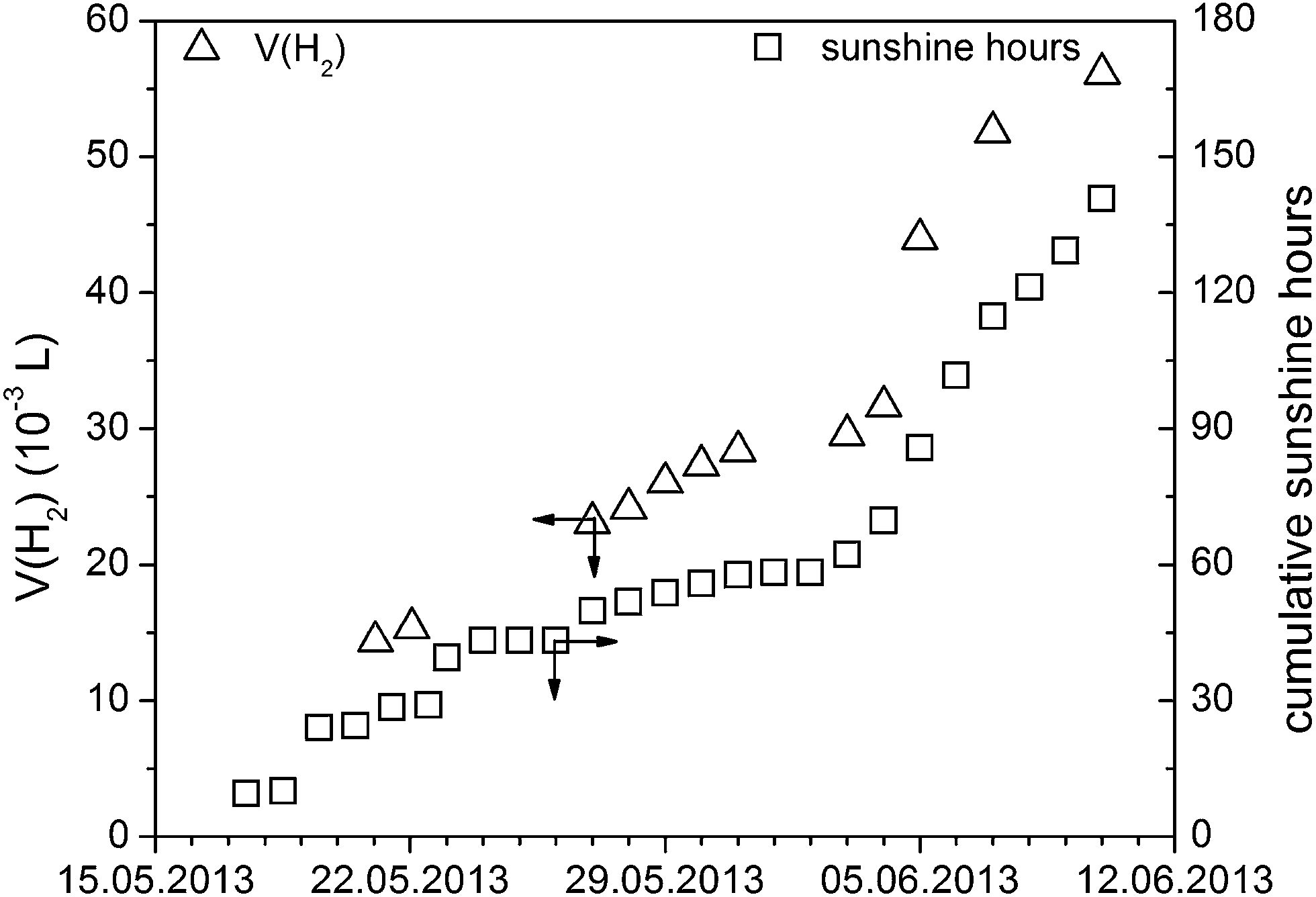 | ||
| Fig. 5 Small scale hydrogen evolution with real sunlight using a small glass reactor equipped with a glass burette for hydrogen quantification (see Fig. S5†) (1.3 g L−1ex situ Pt@mp-CN, 2.5 wt% Pt related to mp-CN, 10 vol% SA, room temperature, real sunlight) which is shown in figure that should run with mp-CN as sustainable photocatalyst. | ||
 | ||
| Fig. 6 Large scale hydrogen evolution with real sunlight using a newly designed photoreactor setup which consists of (a) a reservoir for catalyst dispersion, (b) a peristaltic pump to keep the catalyst dispersed and (c) the photoreactor with an irradiation area of about 1 m2 (0.62 g L−1ex situ loaded mp-CN, ambient conditions, real sunlight). | ||
In our kinetic studies with mp-CN we found that in situ co-catalyst loading should be avoided for a better utilization of the expensive noble metals, e.g. Pt, and under optimized conditions more hydrogen can be produced only by having a larger irradiation area. Therefore, we investigated the ex situ co-catalyst loading via microemulsion in higher amounts (2 g scale) which is an important aspect for the scale-up where higher amounts of this photocatalyst are needed. Furthermore, we increased the irradiation area from about 12 cm2 in lab scale to about 1 m2 for the large scale photoreactor. This corresponds to a scale-up factor of 800 with respect to the irradiation area and the experiments were performed with catalyst dispersions.
With the large scale setup, two experiments with real sunlight were performed. It should be mentioned that for the calculations only the sun hours were considered. The first experiment was done directly after charging the setup with the photocatalyst, water and TEOA, and we obtained a hydrogen evolution rate of about 0.08 L h−1 m−2 (80 mL h−1) for a period of one day. The second experiment was done one month later and we obtained a hydrogen evolution rate of about 0.04 L h−1 m−2 (40 mL h−1) for a period of three days. In comparison to the lab scale experiment, we produced 80 to 160 times more hydrogen, but the rate was lower. We could identify three main reasons for the lower activity. The first reason is the dispersion itself. In the experiment we used a lower catalyst concentration (0.62 g L−1 instead of 1.3 g L−1) so that based on our earlier investigations the rate must be lower (approximately 0.21 L h−1 m−2).22 The second reason is that it was not possible to keep the catalyst dispersed all the time. A larger fraction of the catalyst settled down and could not be re-dispersed by the pump. The third reason is the used polymeric window which was made of polycarbonate (Makrolon). The window absorbs also the UV part of the sun spectrum (shown in Fig. S4†), which was always used in the lab scale experiments with the sun-light simulator. Although, mp-CN is able to absorb light in the visible range, the natural UV fraction leads to a higher photocatalytic activity in hydrogen evolution reaction. Nevertheless, regardless of these minor issues, the first runs with this large scale setup successfully show the hydrogen evolution for mp-CN in the real sunlight conditions. However, modifications of the large scale setup are necessary to obtain the expected rate that was lowered by a factor of 2.5 to 5 due to catalyst settling and increased light absorption by the polycarbonate window.
4. Conclusion
A new ex situ method based on microemulsions was investigated for loading various metals as co-catalyst on porous carbon nitrides. We found that the particle size and distribution is optimized by applying this method for loading Pt on porous carbon nitrides and the effective platinum loading is about 10 times higher in comparison to in situ photodeposited Pt. Moreover, the hydrogen evolution rate was improved by about 40%, which led to the assumption that the better distribution of the nanoparticles by the microemulsion approach leads to increased photocatalytic activity. Furthermore, the microemulsion approach allows for the synthesis of larger amounts of co-catalyst loaded carbon nitrides, which were used for large scale hydrogen evolution with real sunlight in a newly designed photoreactor setup having an irradiation area of 1 m2. In comparison to the lab scale experiments the total amount of produced hydrogen was increase by a factor of 80 to 160 (40 to 80 mL h−1). However, further modifications are in progress, in which the Pt@mp-CN catalyst is immobilized on conductive plates without leaching for long-term use and further the window will be exchanged for better UV transmittance.Acknowledgements
This work was supported by the BMBF (Spitzenforschung und Innovation in den neuen Ländern, FKZ 03IS2071D).References
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, Graphitic Carbon Nitride Materials: Variation of Structure and Morphology and Their Use as Metal-Free Catalysts, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- B. V. Lotsch and W. Schnick, Thermal Conversion of Guanylurea Dicyanamide into Graphitic Carbon Nitride via Prototype CN X Precursors, Chem. Mater., 2005, 17, 3976–3982 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, A Metal-Free Polymeric Photocatalyst for Hydrogen Production from Water under Visible Light, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Polymeric Graphitic Carbon Nitride as a Heterogenous Organocatalyst: From Photochemistry to Multipurpose Catalysis to Sustainable Chemistry, Angew. Chem., Int. Ed., 2011, 51, 68–89 CrossRef CAS PubMed.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, Polymer Semiconductors for Artificial Photosynthesis: Hydrogen Evolution by Mesoporous Graphitic Carbon Nitride with Visible Light, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- A. Thomas, Functional Materials: From Hard to Soft Porous Frameworks, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed.
- K. Kailasam, J. D. Epping, A. Thomas, S. Losse and H. Junge, Mesoporous Carbon Nitride–Silica Composites by a Combined Sol–Gel/thermal Condensation Approach and Their Application as Photocatalysts, Energy Environ. Sci., 2011, 4, 4668–4674 CAS.
- Y. Wang, X. Wang, M. Antonietti and Y. Zhang, Facile One-Pot Synthesis of Nanoporous Carbon Nitride Solids by Using Soft Templates, ChemSusChem, 2010, 3, 435–439 CrossRef CAS PubMed.
- X. Chen, Y.-S. Jun, K. Takanabe, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Ordered Mesoporous SBA-15 Type Graphitic Carbon Nitride: A Semiconductor Host Structure for Photocatalytic Hydrogen Evolution with Visible Light, Chem. Mater., 2009, 21, 4093–4095 CrossRef CAS.
- J. Zhang, G. Zhang, X. Chen, S. Lin, L. Möhlmann, G. Dołęga, G. Lipner, M. Antonietti, S. Blechert and X. Wang, Co-Monomer Control of Carbon Nitride Semiconductors to Optimize Hydrogen Evolution with Visible Light, Angew. Chem., Int. Ed., 2012, 51, 3183–3187 CrossRef CAS PubMed.
- Y.-S. Jun, E. Z. Lee, X. Wang, W. H. Hong, G. D. Stucky and A. Thomas, From Melamine-Cyanuric Acid Supramolecular Aggregates to Carbon Nitride Hollow Spheres, Adv. Funct. Mater., 2013, 23, 3661–3667 CrossRef CAS.
- Y.-S. Jun, J. Park, S. U. Lee, A. Thomas, W. H. Hong and G. D. Stucky, Three-Dimensional Macroscopic Assemblies of Low-Dimensional Carbon Nitrides for Enhanced Hydrogen Evolution, Angew. Chem., Int. Ed., 2013, 52, 11083–11087 CrossRef CAS PubMed.
- K. Kailasam, J. Schmidt, H. Bildirir, G. Zhang, S. Blechert, X. Wang and A. Thomas, Room Temperature Synthesis of Heptazine-Based Microporous Polymer Networks as Photocatalysts for Hydrogen Evolution, Macromol. Rapid Commun., 2013, 34, 1008–1013 CrossRef CAS PubMed.
- L. Ge, C. Han and J. Liu, In Situ Synthesis and Enhanced Visible Light Photocatalytic Activities of Novel PANI–g-C3N4 Composite Photocatalysts, J. Mater. Chem., 2012, 22, 11843–11850 RSC.
- J. Zhang, J. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Fu and X. Wang, Sulfur-Mediated Synthesis of Carbon Nitride: Band-Gap Engineering and Improved Functions for Photocatalysis, Energy Environ. Sci., 2011, 4, 675–678 CAS.
- Y. Wang, J. Zhang, X. Wang, M. Antonietti and H. Li, Boron- and Fluorine-Containing Mesoporous Carbon Nitride Polymers: Metal-Free Catalysts for Cyclohexane Oxidation, Angew. Chem., Int. Ed., 2010, 49, 3356–3359 CrossRef CAS PubMed.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, A Facile Approach to Synthesize Novel Oxygen-Doped g-C3N4 with Superior Visible-Light Photoreactivity, Chem. Commun., 2012, 48, 12017–12019 RSC.
- G. Dong, K. Zhao and L. Zhang, Carbon Self-Doping Induced High Electronic Conductivity and Photoreactivity of g-C3N4, Chem. Commun., 2012, 48, 6178–6180 RSC.
- M. Schwarze, D. Stellmach, M. Schröder, K. Kailasam, R. Reske, A. Thomas and R. Schomäcker, Quantification of Photocatalytic Hydrogen Evolution, Phys. Chem. Chem. Phys., 2013, 15, 3466–3472 RSC.
- M. Schwarze, M. Schröder, D. Stellmach, K. Kailasam, A. Thomas and R. Schomäcker, Entwicklung Eines Reaktors Zur Standardisierten Quantifizierung Der Photokatalytischen Wasserstofferzeugung, Chem. Ing. Tech., 2013, 85, 500–507 CrossRef CAS.
- D. Hollmann, M. Karnahl, S. Tschierlei, K. Kailasam, M. Schneider, J. Radnik, K. Grabow, U. Bentrup, H. Junge and M. Beller, et al. Structure–Activity Relationships in Bulk Polymeric and Sol–Gel-Derived Carbon Nitrides during Photocatalytic Hydrogen Production, Chem. Mater., 2014, 26, 1727–1733 CrossRef CAS.
- M. Schröder, K. Kailasam, S. Rudi, M. Richter, A. Thomas, R. Schomäcker and M. Schwarze, Impact of the Reaction Conditions on the Photocatalytic Reduction of Water on Mesoporous Polymeric Carbon Nitride under Sunlight Irradiation, Int. J. Hydrogen Energy, 2014, 39, 10108–10120 CrossRef PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Chemical Synthesis of Mesoporous Carbon Nitrides Using Hard Templates and Their Use as a Metal-Free Catalyst for Friedel–Crafts Reaction of Benzene, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- X.-H. Li, J. Zhang, X. Chen, A. Fischer, A. Thomas, M. Antonietti and X. Wang, Condensed Graphitic Carbon Nitride Nanorods by Nanoconfinement: Promotion of Crystallinity on Photocatalytic Conversion, Chem. Mater., 2011, 23, 4344–4348 CrossRef CAS.
- J. Yu, S. Wang, B. Cheng, Z. Lin and F. Huang, Noble Metal-Free Ni(OH)2–g-C3N4 Composite Photocatalyst with Enhanced Visible-Light Photocatalytic H2-Production Activity, Catal. Sci. Technol., 2013, 3, 1782–1789 CAS.
- J. Dong, M. Wang, X. Li, L. Chen, Y. He and L. Sun, Simple Nickel-Based Catalyst Systems Combined with Graphitic Carbon Nitride for Stable Photocatalytic Hydrogen Production in Water, ChemSusChem, 2012, 5, 2133–2138 CrossRef CAS PubMed.
- X. Zhou, Z. Luo, P. Tao, B. Jin, Z. Wu and Y. Huang, Facile Preparation and Enhanced Photocatalytic H2-Production Activity of Cu(OH)2 Nanospheres Modified Porous g-C3N4, Mater. Chem. Phys., 2014, 143, 1462–1468 CrossRef CAS PubMed.
- L. Ge, C. Han, X. Xiao and L. Guo, In Situ Synthesis of Cobalt–phosphate (Co–Pi) Modified g-C3N4 Photocatalysts with Enhanced Photocatalytic Activities, Appl. Catal., B, 2013, 142–143, 414–422 CrossRef CAS PubMed.
- Y. Di, X. Wang, A. Thomas and M. Antonietti, Making Metal-Carbon Nitride Heterojunctions for Improved Photocatalytic Hydrogen Evolution with Visible Light, ChemCatChem, 2010, 2, 834–838 CrossRef CAS.
- L. Ge, C. Han, J. Liu and Y. Li, Enhanced Visible Light Photocatalytic Activity of Novel Polymeric G-C3N4 Loaded with Ag Nanoparticles, Appl. Catal., A, 2011, 409–410, 215–222 CrossRef CAS PubMed.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, Photocatalytic Activities of Graphitic Carbon Nitride Powder for Water Reduction and Oxidation under Visible Light, J. Phys. Chem. C, 2009, 113, 4940–4947 CAS.
- H. Ha, J. Be, A. Palmqvist, M. Skoglundh, H. Qiuhong and L. K. L. Falk, Deposition of Platinum Nanoparticles, Synthesized in Water-in-Oil Microemulsions, on Alumina Supports, Langmuir, 2002, 18, 1811–1818 CrossRef.
- R. Y. Parapat, V. Parwoto, M. Schwarze, B. Zhang, D. S. Su and R. Schomäcker, A New Method to Synthesize Very Active and Stable Supported Metal Pt Catalysts: Thermo-Destabilization of Microemulsions, J. Mater. Chem., 2012, 22, 11605–11614 RSC.
- R. Y. Parapat, M. Wijaya, M. Schwarze, S. Selve, M. Willinger and R. Schomäcker, Particle Shape Optimization by Changing from an Isotropic to an Anisotropic Nanostructure: Preparation of Highly Active and Stable Supported Pt Catalysts in Microemulsions, Nanoscale, 2013, 5, 796–805 RSC.
- G. Ertl, H. Knozinger and J. Weitkamp, Handbook of Heterogeneous Catalysis, 2008, Wiley–VCH Search PubMed.
- Y. Wang, J. Hong, W. Zhang and R. Xu, Carbon Nitride Nanosheets for Photocatalytic Hydrogen Evolution: Remarkably Enhanced Activity by Dye Sensitization, Catal. Sci. Technol., 2013, 3, 1703–1711 CAS.
- S. Deguchi, T. Takeichi, S. Shimasaki and M. Ogawa, Photocatalytic Hydrogen Production from Water with Nonfood Hydrocarbons as Oxidizing Sacrifice Agents, AIChE J., 2011, 57, 2237–2243 CrossRef CAS.
- R. Y. Parapat, V. Parwoto, M. Schwarze, B. Zhang, D. S. Su and R. Schomäcker, A New Method to Synthesize Very Active and Stable Supported Metal Pt Catalysts: Thermo-Destabilization of Microemulsions, J. Mater. Chem., 2012, 22, 11605 RSC.
- Q. Li, J. Zhang, Z. Jin, C. Feng, J. Zhang, Z. Wu and Z. Zhang, A Novel TiO2 with a Large Amount of Bulk Intrinsic defects—Visible-Light-Responded Photocatalytic Activity Induced by Foreign Trap, Chin. Sci. Bull., 2013, 58, 1675–1681 CrossRef CAS.
- Y. Li, K. Zhang, S. Peng, G. Lu and S. Li, Photocatalytic Hydrogen Generation in the Presence of Ethanolamines over Pt/ZnIn2S4 under Visible Light Irradiation, J. Mol. Catal. A: Chem., 2012, 363–364, 354–361 CrossRef CAS PubMed.
- H. W. Kang and S. B. Park, H2 Evolution under Visible Light Irradiation from Aqueous Methanol Solution on SrTiO3:Cr/Ta Prepared by Spray Pyrolysis from Polymeric Precursor, Int. J. Hydrogen Energy, 2011, 36, 9496–9504 CrossRef CAS PubMed.
- K. Maeda, H. Terashima, K. Kase and K. Domen, Nanoparticulate Precursor Route to Fine Particles of TaON and ZrO2–TaON Solid Solution and Their Photocatalytic Activity for Hydrogen Evolution under Visible Light, Appl. Catal., A, 2009, 357, 206–212 CrossRef CAS PubMed.
- F. Gärtner, A. Boddien, E. Barsch, K. Fumino, S. Losse, H. Junge, D. Hollmann, A. Brückner, R. Ludwig and M. Beller, Photocatalytic Hydrogen Generation from Water with Iron Carbonyl Phosphine Complexes: Improved Water Reduction Catalysts and Mechanistic Insights, Chem.–Eur. J., 2011, 17, 6425–6436 CrossRef PubMed.
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Photocatalytic Hydrogen and Oxygen Formation under Visible Light Irradiation with M-Doped InTaO4 (M = Mn, Fe, Co, Ni and Cu), J. Photochem. Photobiol., A, 2002, 148, 65–69 CrossRef CAS.
- S.-C. Yu, C.-W. Huang, C.-H. Liao, J. C. S. Wu, S.-T. Chang and K.-H. Chen, A Novel Membrane Reactor for Separating Hydrogen and Oxygen in Photocatalytic Water Splitting, J. Membr. Sci., 2011, 382, 291–299 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra10814k |
| This journal is © The Royal Society of Chemistry 2014 |