
Recent developments in Ritter reaction
Abstract
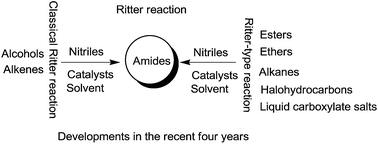
Ritter reaction is an atom economy reaction which produces an amide through the reaction of a nitrile with an alcohol or alkene in the presence of an acid. A number of important advances have been achieved in recent years with respect to substrates, the variety of catalysts, the reaction media and the diversity of products. This paper reviews recent findings and assesses the Ritter reaction.
 Please wait while we load your content...
Please wait while we load your content...

