
Synthesis of aluminum-modified 3D mesoporous TUD-1 materials and their hydrotreating performance of FCC diesel†
Zugang Wang‡
,
Jianye Fu‡,
Yunchuan Deng,
Aijun Duan*,
Zhen Zhao*,
Guiyuan Jiang,
Jian Liu,
Yuechang Wei and
Suoqi Zhao
State Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing 102249, P. R. China. E-mail: duanaijun@cup.edu.cn; zhenzhao@cup.edu.cn
First published on 28th November 2014
Abstract
Aluminum-modified 3D mesoporous TUD-1 materials (denoted as Al-TUD-1, abbreviated as AT) were successfully synthesized by a sol–gel method using TEA and TEAOH as the co-template. Several analytical techniques such as XRD, N2 physisorption, TEM, NH3-TPD, H2-TPR and UV-vis DRS were used to characterize the typical physico-chemical properties of Al2O3–AT composite supports and their corresponding catalysts. The characterization results indicated that most of the aluminum species was partially incorporated into the framework of the TUD-1 silicate network, and the as-synthesized material had a uniform pore size distribution, high surface area and high pore volume. The catalytic performances of hydrotreating were evaluated by using PetroChina Hohhot petrochemical company FCC diesel as the feedstock. Among the series catalysts of NiMo/Al2O3–AT-m, the catalyst with 30 m% AT in the support exhibited the highest HDS conversion, which was as high as 97.42%. Moreover, the influence factors in the synthetic process of the Al modified TUD-1 such as Si/Al ratio, heat treatment time and heat temperature were systematically studied and the corresponding supported NiMo series catalysts showed good hydrotreating performances. The catalyst with the Si/Al ratio of 50 in AT, heat treatment time of 18 h and heat treatment temperature of 180 °C exhibited the highest HDS (97.56%) and HDN (99.63%) efficiencies, respectively.
1. Introduction
Diesel is one of the important transportation fuels and diesel engines are 25–40% more fuel-efficient than comparable gasoline engines. Nevertheless, the stringent regulation of exhaust emission consequentially requests ultra clean diesels; moreover, the demand of diesel increases sharply, resulting in the correlative requirements of diesel fuels in quality and quantity.1–4 The sulfur content of transportation fuels is expected to be very low due to the strict fuel specification.5,6 The current efficient technique to remove sulfur from diesel fuel is hydrodesulfurization (HDS). However, it is a great challenge to achieve ultra deep HDS by the conventional HDS processes under typical operation conditions; thus, the development of novel hydrotreating technology is significant for refineries.7 Furthermore, catalysts perform an important role in transforming poor quality diesel into an ultraclean final product. A hydrotreating catalyst with high activity and good selectivity should possess an open pore structure and suitable acidity, and also combine the suitable dispersions of active metals in the catalyst surface with adjustable redox properties for the deep HDS of diesel.Mesoporous materials retain the superiority of large pore sizes, which favors the diminishing of diffusion resistance, and recently they are considered to be the ideal support candidates for HDS catalysts.8 From the first publication of mesoporous MCM-41 in 1992,9 various kinds of mesoporous materials have been extensively studied by scientific researchers from different fields. While the concerns of a refinery focus on the economical routes to synthesize inexpensive porous materials. Thus, non-surfactant templating routes are of great significant to develop and design new types of materials for various catalysis processes. Recently, TUD-1 (Technische Universiteit Delft) and its related materials prepared through low-cost synthesis routes showed the attractive characteristics of high surface area, thick mesopore wall, three-dimensional sponge-like mesoporous substrate accessibility and high stability, which make it a potential candidate to be a support and catalyst.10,11 The purely siliceous TUD-1 may be allocated with Brönsted and Lewis acidities by incorporating metals such as Al into the framework through sol–gel techniques.11–15 Bala and coworkers16 synthesized a series of catalysts, which used TUD-1 and its related materials as supports and noble metals (such as palladium, platinum, ruthenium and other metals) as the hydrogenation active metals for a distillate hydrocarbon feed. From the patent report, the Pt/TUD-1 catalyst showed a high activity of aromatic saturation. Liu17 prepared HPMo/TUD-1 catalysts, which showed a superior activity, high isomer selectivity and excellent long-term stability in the hydroisomerization of alkanes such as n-heptane. Shan18 made a series of NiMo/Al-TUD-1 catalysts and took FCC clarified slurry oil as the feedstock to evaluate their hydrotreating activities. The as-synthesized catalyst exhibited higher activity than the commercial γ-Al2O3 catalyst, while the efficiencies of nitrogen and sulfur removal were 64% and 91%, respectively. Therefore, the mesoporous TUD-1 materials were believed to be an alternative support additive in the hydrotreating process.
In this research, aluminum-modified (three-dimensional) 3D TUD-1 mesoporous materials (noted as Al-TUD-1, abbreviated as AT) were in situ synthesized by a sol–gel method using triethanolamine (TEA) and tetraethyl ammonium hydroxide (TEAOH) as the co-template. Then, a series of catalyst additives with different AT contents (expressed as AT-m) and various Si/Al ratios (represented as AT-r) were prepared by the incipient-wetness impregnation method. Moreover, the synthesis conditions such as heat treatment time and heat treatment temperature were optimized, and the related additives were denoted as AT-ti and AT-te. The catalytic performances were evaluated by using PetroChina Hohhot petrochemical company FCC diesel fuel.
2. Experimental
2.1 Preparation of materials
Al-TUD-1(AT) was synthesized by using TEA as a template in a one-pot surfactant-free procedure based on the sol–gel technique.10,13 AT with various Si/Al ratios can be composed by adjusting the molar ratio of SiO2:xAl2O3:0.4 tetraethylammonium hydroxide (TEAOH):0.5TEA:10H2O. In a typical synthetic process, taking the material with an Si/Al ratio of 25 as an example, 0.68 g of aluminum(III) isopropoxide (≥98%, Sinopharm Chemical Reagent Co., Ltd (SCRC), China) was dissolved in 20 mL ethanol solution, and then the above mixture was added dropwise to 17.30 g of tetraethyl orthosilicate solution (TEOS, ≥98%, SCRC). After stirring for a few minutes, 12.50 g of TEA (≥97%, SCRC) was added dropwise into the mixture. When the solution was in a homogeneous phase, a mixture of 5.32 g of water and 14.28 g of TEAOH (25 wt% aqueous solution, Hangzhou Greenda Chemical Co., Ltd, China) was added under vigorous stirring. A clear gel was obtained after these steps, then it needed to be aged at room temperature for 24 h and dried at 100 °C for 10 h, followed by a hydrothermal treatment in a Teflon-lined autoclave at 180 °C for 18 h, and finally calcined in air at 600 °C for 10 h to remove the organic compounds. By using the same method, a series of different AT composites with various Si/Al ratios, heat treatment times and heat treatment temperatures were synthesized. The as-synthesized supports were obtained by mechanically mixing AT and γ-Al2O3, and the supports with different AT proportions were denoted as AT-m, where m represents the AT proportion in the support. Moreover, the as-synthesized materials obtained at different conditions, involving various Si/Al ratios, heat treatment times and heat treatment temperatures, were represented as AT-r, AT-ti and AT-te, where r represents the Si/Al ratio, ti represents heat treatment time and te represents heat treatment temperature in the AT composite, respectively.2.2 Preparation of supports and corresponding catalysts
The mechanical mixture of Al2O3 and AT (termed as Al2O3–AT) was impregnated by Ni and Mo species to prepare the supported catalysts through a two-step incipient-wetness impregnation method with the aqueous solutions of ammonium heptamolybdate ((NH4)6Mo7O24·4H2O, ≥99%, SCRC) and nickel nitrate (Ni(NO3)2·6H2O, ≥99%, SCRC). After each impregnation step, the prepared precursors were dispersed in an ultrasonic bath for 30 min. The prepared samples were dried at 110 °C for 4 h, and calcined at 550 °C for 4 h. The corresponding supported catalysts had a final composition of 81 wt% support, and active metal contents of MoO3 15.5 wt%, and NiO 3.5 wt%. The AT additive contents were varied during this study. In other cases, the support typically contained 70 wt% γ-Al2O3 and 30 wt% AT. Moreover, the corresponding catalysts were noted as NiMo/Al2O3–AT-m (r, ti, or te). The traditional NiMo/Al2O3 catalyst was taken as the reference catalyst to evaluate the catalytic performance.2.3 Characterization of supports and catalysts
Nitrogen adsorption–desorption isotherms were measured at −196 °C on a static volumetric instrument Autosorb-6b (Quanta Chrome). Prior to each measurement, the sample was degassed at 250 °C for 12 h under high vacuum. The specific surface area was estimated by the Brunauer–Emmett–Teller (BET) method,19 and the pore size distribution was calculated by the Barrett–Joyner–Halenda (BJH) method20 using the adsorption isotherm branch.Powder X-ray diffraction (XRD) patterns were recorded on a Bruker AXS D8 diffractometer (under ambient conditions) using filtered Cu Kα radiation (λ = 0.15406 nm) operated at 40 kV and 40 mA. Diffraction data were collected from 0.6° to 60° with a resolution of 0.01° (2θ).
The surface morphology of the catalyst was observed by scanning electron microscopy (SEM) on a Quanta 200F instrument using an accelerating voltage of 5 kV in combination with an EDAX genesis 4000 energy-dispersive X-ray spectrometer (EDS). The samples for SEM were dusted on an adhesive conductive carbon belt attached to a copper disk and were coated with 10 nm Au before measurement.
Transmission electron microscopy (TEM) was performed on a JEM 2100 LaB6 (JEOL) equipment with an accelerating voltage of 300 kV. A few mg of the powdered samples was suspended in 2 mL ethanol, and the suspension was treated in a sonicator for 1 h. Then, the suspension was left to settle for 15 min before a drop was taken and dispersed on a 300 mesh carbon-coated grid. 27Al solid-state magic-angle-spinning nuclear magnetic resonance (27Al MAS NMR) spectra were recorded on a Bruker MSL-300NMR spectrometer with the frequency at 59.6 MHz.
The UV-vis diffuse reflectance spectra (DRS) experiments were performed on a Hitachi U-4100 UV-vis spectrophotometer with an integration sphere diffuse reflectance attachment. The powder samples were loaded in a transparent quartz cell and were measured in the region of 200–800 nm at room temperature. BaSO4 reflectance was used as the baseline for the corresponding sample measurement.
Acid properties of different materials were characterized by temperature-programmed desorption of ammonia (NH3-TPD) method and pyridine-adsorbed Fourier transformed infrared spectroscopy (Py-FTIR). 0.1 g sample was pretreated in helium at 550 °C for 4 h, then cooled to 100 °C and adsorbed NH3 for 30 min. After flushing by pure helium gas at 100 °C for 1 h, temperature-programmed desorption started at a rate of 10 °C min−1 from 100 to 550 °C, and the signal was monitored by a thermal conductivity detector (TCD). The nature of the acid sites was tested by Py-FTIR experiments using a MAGNAIR 560 FTIR instrument (Nicolet Co., USA) with the resolution of 1 cm−1. The samples were dehydrated at 500 °C for 5 h under a vacuum of 1.33 × 10−3 Pa, followed by the adsorption of purified pyridine vapor at room temperature for 20 min. Then, the system was evacuated at different temperatures and the pyridine-adsorbed IR spectra were recorded.
H2-TPR was tested on a self-assembled instrument containing 10% hydrogen in argon at a constant flow rate of 40 mL min−1 and the heating rate of 10 °C min−1. The system was then degassed and evacuated at different temperatures, and finally the IR spectra were recorded. The absolute amount of pyridine adsorbed on the Brönsted acid sites and Lewis acid sites can be calculated using IMEC(B) = 1.67 cm μmol−1, and IMEC(L) = 2.22 cm μmol−1 described by Emeis.21,22
2.4 Catalytic activity measurement
The feedstock used in this research was PetroChina Hohhot petrochemical company FCC diesel with a S content of 1013.8 mg L−1 and N content of 640.3 mg L−1. The properties of the diesel oil are shown in Table 1. A definite amount of catalyst (grain size of 40–60 mesh) was loaded to evaluate the catalytic performances in a high-pressure fixed-bed reactor. All of the catalysts were presulfided by a 2 m% CS2–cyclohexane and H2 mixture for 4 h at 320 °C and 4 MPa.| Properties | Data | Properties | Data | |
|---|---|---|---|---|
| a IBP and FBP are the abbreviations for initial boiling point and final boiling point, respectively. | ||||
| N, mg L−1 | 640.3 | Engler distillation (°C) | IBP | 144 |
| S, mg L−1 | 1013.8 | 5% | 177 | |
| Density @ 20 °C, g cm−3 | 0.8686 | 10% | 191 | |
| Hydrocarbon type | 30% | 215 | ||
| Saturates (v%) | 39.3 | 50% | 240 | |
| Aromatics (v%) | 49.0 | 70% | 272 | |
| Olefin (v%) | 11.7 | 90% | 310 | |
| Cetane number | 37.7 | 95% | 325 | |
| FBP | 326 | |||
After presulfidation, the temperature was increased to the reaction temperature of 350 °C, while the pressure was 5.0 MPa, H2 to oil ratio was 600 mL per mL and LHSV was 1.0 h−1. The catalytic activities under investigation were evaluated by HDS and HDN efficiencies. The total sulfur and nitrogen contents in the feed and products were measured by a RPP-2000SN sulfur analyzer system (Taizhou Central Analytical Instruments Co., Ltd, China). The HDS and HDN efficiency is defined as follows:
| HDS% efficiency = [(Sf − Sp)/Sf] × 100% |
| HDN% efficiency = [(Nf − Np)/Nf] × 100% |
3. Results and discussion
3.1 Characterization of the materials
Fig. 1 shows the powder X-ray diffraction (XRD) patterns of AT-r (r equals to 25, 50, 75, 100, and 200). From the small angle domain in Fig. 1a, one intense peak can be found at a low angle (2θ = 0.7°–1.2°), indicating that the materials possess a mesoporous structure. From the wide angle domain in Fig. 1b, the very broad peak centered around 2θ of 24° is attributed to the amorphous silica and no apparent signals of Al2O3 crystalline phases are detected; therefore, it seems that the Al species are highly dispersed in the framework of Si or in the amorphous non-framework phases. Simultaneously, the characteristic peaks also exhibit an intensity increase enhancement in Si/Al ratios, which is ascribed to the uniform packing of nano-sized particles.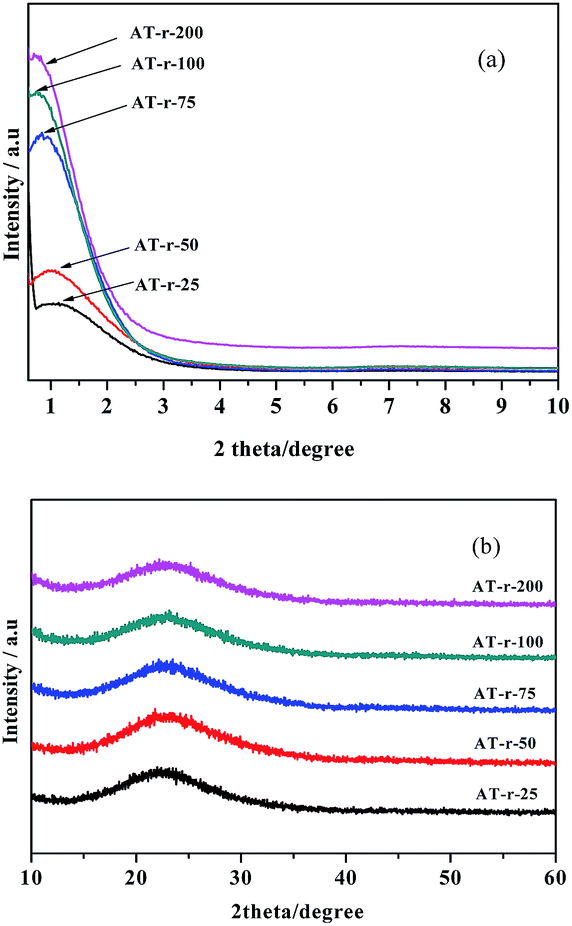 | ||
| Fig. 1 X-Ray diffractions of the AT-r materials. (a) Low-angle patterns, (b) wide-angle patterns. Notes: all samples were heat-treated under 180 °C for 18 h. | ||
As shown in Fig. 2, the transmission electron microscopy (TEM) images of the AT-r materials display a sponge-like or worm-like mesoporous network structure, which is similar to the pure silicous TUD-1 mesoporous materials.10 No evidence of alumina or other phases can be detected in the patterns, which is consistent with the XRD results, suggesting that aluminum might be incorporated into the framework or the sizes of the isolated aluminum are small and beyond XRD detection limitation.10,12,13 Moreover, the physical mixture of γ-Al2O3 and AT-r-50 was also characterized by TEM as shown in Fig. S1,† and the result showed that this mixture clearly consisted of two phases due to the mechanical mixing.
 | ||
| Fig. 2 TEM images of AT-r materials. (a) & (b) AT-r-25; (c) AT-r-50; (d) AT-r-75; (e) AT-r-100; (f) AT-r-200. | ||
Scanning electron microscopy (SEM) study was carried out to investigate the surface morphology of the AT-r materials. The micrographs of AT-r-25, AT-r-50, AT-r-75, AT-r-100 and AT-r-200 are very similar to each other; and they all show the non-irregular morphology of the TUD-1 particles as described by others.23 It is evident that the morphology of the AT materials is virtually unchanged after aluminum incorporation. The square regions in Fig. S2a–e† are the analysis positions; furthermore, the EDX results collected from different materials showed similar Si/Al ratios as the chemical compositions of the synthesis solution.
The textural properties of the AT-r samples obtained from the N2 adsorption–desorption studies are shown in Fig. 3, which exhibit the characteristics of type IV isotherms (Fig. 3a) according to the IUPAC classification, indicating that these materials have mesoporous features24 and narrow pore-size distributions25 (Fig. 3b). The presence of mesopores is evident from the uptake of nitrogen at a relative pressure (P/P0) of 0.4–0.8 as a result of nitrogen condensation inside the mesopores. The difference between the adsorption and desorption isotherms proves the presence of percolation or network effects in the pores. The closure of the hysteresis loop at P/P0 values between 0.4 and 0.45 for all the AT-r samples can be explained by the tensile strength effect, which demonstrates that the samples have relatively narrow pore size distributions within 5 to 7 nm.25
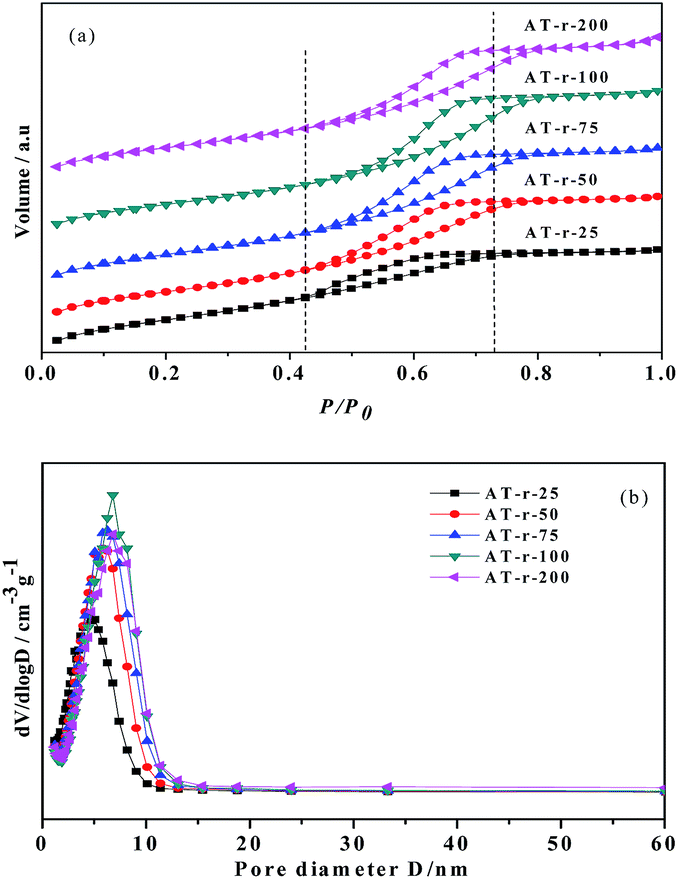 | ||
| Fig. 3 The N2 adsorption–desorption isotherms (a) and the pore size distributions (b) of the AT-r samples. | ||
Similar to the AT-r samples, the textural properties of the AT-ti samples obtained from the N2 sorption studies are shown in Fig. 4, and all samples show the typical mesoporous material characteristics (Fig. 4a); moreover, their pore size distributions (Fig. 4b) are relatively narrow. Furthermore, the pore diameters enlarge with increase in heating time in the synthetic process.
 | ||
| Fig. 4 The N2 adsorption–desorption isotherms (a) and the pore size distributions (b) of the AT-ti samples. Notes: AT-ti was synthesized with Si/Al = 50 and heat treated under 180 °C. | ||
Tables 2 and 3 are the sum of the surface areas, calculated from the absorption branch of the N2 adsorption–desorption isotherms using the Brunauer–Emmett–Teller (BET) method, whereas the pore volume and pore diameter are calculated from the adsorption branch of the N2 adsorption–desorption isotherms using the Barrett–Joyner–Halenda method. From the data in Tables 2 and 3, it can be found that the surface areas of the samples range from 513 to 759 m2 g−1, the pore volumes are from 0.4 to 1.0 cm3 g−1 and the mesopore diameters are estimated from 4 to 8 nm. Compared with γ-Al2O3, the as-synthesized samples have higher surface areas and narrower pore size distributions. Note that the N2 adsorption results coincide with the TEM analysis.
| Materials | Si/Al ratio | SBET (m2 g−1) | VBJH (cm3 g−1) | Pore diameter (nm) |
|---|---|---|---|---|
| AT-r-25 | 25 | 732 | 0.77 | 4.7 |
| AT-r-50 | 50 | 728 | 0.91 | 5.8 |
| AT-r-75 | 75 | 759 | 0.98 | 6.3 |
| AT-r-100 | 100 | 702 | 0.98 | 6.8 |
| AT-r-200 | 200 | 709 | 0.98 | 6.8 |
| γ-Al2O3 | 0 | 239 | 0.86 | 8.2 |
| Materials | Heat time (h) | SBET (m2 g−1) | VBJH (cm3 g−1) | Pore diameter (nm) |
|---|---|---|---|---|
| AT-ti-1 h | 1 | 553 | 0.42 | 3.4 |
| AT-ti-7 h | 7 | 513 | 0.66 | 4.3 |
| AT-ti-12 h | 12 | 681 | 0.85 | 4.3 |
| AT-ti-18 h | 18 | 717 | 0.92 | 4.9 |
| AT-ti-48 h | 48 | 648 | 0.86 | 5.5 |
From the results of Table 3, it can be observed that the pore diameters increase with the extension of the heat treatment time, whereas the pore volumes of the AT-ti materials increase at first and then decrease gradually. This phenomena could be explained by other researches in the interpretation of pore formation mechanism in TUD-1.14 Firstly, TEOS hydrolyses to form Si(OH)4 groups, which condense with TEA. Since C–O–Si bonds are more easily hydrolysed than Si–O–Si bonds, the TEA–silica linkages are repeatedly broken and the silica nuclei grow slowly.
Under the effect of heating, the processes of TEA hydrolysis and silanol condensation are accelerated, leading to a controlled phase separation. Subsequently, TEA aggregates to construct the mesoporous silica framework and the silica species condense around the cores simultaneously. Finally, the TEA aggregates are removed, and the product with foam-like mesoporous porosity is prepared. In this research, sufficient time was needed to balance the self-aggregation process of TEA in a certain range. Thus, the pore diameter, pore volume and specific surface area of the prepared materials increased with the TEA self-aggregation degree. When the heat treatment time was maintained beyond a suitable range (in this research, the suitable heat time was 18 h), the TEA template would have a significant accumulation in the self-assembly process, which resulted in the increase of the pore sizes and the decrease of the pore volumes and specific surface areas. This suggested that the optimized pore structures of the synthetic material could be modulated by adjusting the heat treatment time, which contributed a lot to the better diffusion performance of the macromolecules in the reaction process.
27Al nuclear magnetic resonance (NMR) was used to analyze the existence states of the Al species in the as-synthesized materials. As shown in Fig. 5, the spectra of all the AT-r samples present two distinct peaks at δ = 54 ppm and δ = 0 ppm. The former signal at δ = 54 ppm can be assigned to the aluminum species (AlO4 structural unit) in a tetrahedral framework environment.26–28 The other peak with low intensity at δ = 0 ppm can be attributed to the non-framework Al species (AlO6 structural unit) with octahedral coordination,29,30 which covers about one-third of all the Al species, as described by another researcher.31 The results indicate that most of the Al are incorporated into the TUD-1 framework rather than existing as non-framework Al species because the signals at δ = 0 ppm decrease with increase in the Si/Al ratio.
 | ||
| Fig. 5 27Al MAS NMR spectra of the AT-r materials. | ||
3.2 Characterizations of the supported catalysts
Fig. 6 is the XRD profiles of the supported NiMo catalysts. The characteristic diffraction peaks of γ-Al2O3 (2θ = 37.60°, 45.84°, 67.30°, JCPDS Card no. 10-0425) appear in curves (c–f). Compared with their corresponding supports, NiMo/γ-Al2O3, NiMo/Al2O3–AT-r-50 and NiMo/Al2O3–AT-r-200 catalysts exhibit decreasing intensities after the incorporation of NiMo active metals and AT-r additives with γ-Al2O3. For these catalysts, weak XRD diffraction peaks corresponding to MoO3 (2θ = 23.33°, 25.70°, 27.33°, 33.13°, 33.73°, 38.97°, 39.65°, 46.28°, JCPDS Card no. 05-0508) were found at 2θ = 27.3°; however, there was no diffraction peak belonging to the existing NiO (JCPDS Card no. 78-0429) or NiAl2O4 (JCPDS Card no. 78-0552) crystal phases, which demonstrates that the NiO species is relatively highly dispersed on the catalyst surface. However, the diffraction peak of MoO3 suggests that there is bulk MoO3 formed on the supports (Fig. 6, curves (a–d)), indicating that the support-metal interaction (MSI) in these four catalysts are not as strong as that in the Al2O3 based catalyst, whereas the weak MSI favors the formation of small stacked MoO3 crystals on the support. Moreover, the dispersed MoO3 nanocrystallines with suitable stacks are believed to improve the catalytic activity, since they can provide more effectively brim and edge active centers for the reactant molecules.32 | ||
| Fig. 6 X-Ray diffractions of the NiMo/Al2O3–AT-r catalysts. (a) NiMo/AT-r-25; (b) NiMo/AT-r-200; (c) NiMo/Al2O3–AT-r-25; (d) NiMo/Al2O3–AT-r-200; (e) NiMo/Al2O3; (f) Al2O3. | ||
The textural properties of the NiMo/Al2O3–AT-r catalysts are shown in Table 4. From the data in Table 4, the specific surface areas, pore volumes and pore diameters of the NiMo/Al2O3–AT-r catalysts are higher than that of NiMo/Al2O3. As is well known, a high surface area is adequate for dispersing active metals to form more active sites, whereas a large pore diameter favors to reduce the diffusion hindrance and promotes the accessibility of the active sites for the reactant molecules. The N2 adsorption–desorption isotherms of NiMo/AT-r and NiMo/AT-ti are shown in Fig. S3 and S4.† The textural properties of the above two catalysts are shown in Tables S1 and S2.† All the isotherms remained type IV isotherms, indicating that the impregnations of active metal species did not influence the pore structure greatly, whereas the SBETs and pore diameters decreased after impregnation possibly due to the coverage of metal depositions and the blockage of micro channels.
| Catalyst number | SBET (m2 g−1) | VBJH (cm3 g−1) | Pore diameter(nm) |
|---|---|---|---|
| NiMo/Al2O3–AT-r-50 | 183 | 0.43 | 9.4 |
| NiMo/Al2O3–AT-r-200 | 198 | 0.44 | 8.8 |
| NiMo/Al2O3 | 167 | 0.34 | 8.3 |
UV-vis DRS was applied to analyze the local structures concerning the Mo oxide species on the supports. Hence, the adsorption bands observed in the spectra are produced only by the ligand-to-metal charge transfer O2− → Mo6+. The positions of the absorption bands are determined by the coordination and agglomeration degrees of the Mo species in the samples. Fig. S5† shows the UV-vis DRS spectra of the NiMo/Al2O3–AT-r series catalysts in the range from 200 to 800 nm based on the backgrounds of the supports. The band at 220–250 nm is commonly attributed to the isolated tetrahedral molybdate. The band at 280 nm is assigned to monomer, dimer or oligomer molybdate species, whereas the band at 280–320 nm is allocated to the Mo–O–Mo bridge bond of the octahedral coordination Mo species, and the bulk MoO3 absorbs at 300–330 nm.33–35 Moreover, no obvious broad bands of NiO and NiMoO4 are found at 330–350 nm and 725–740 nm.36 The red shift of the lowest energy transition absorption of molybdate indicates that the molybdate species exist in a high dispersion phase and the particle sizes of the MoO3 crystals are very small, which is consistent with the result of XRD characterization.
NH3-TPD was performed to characterize the surface acidity of the catalysts. NH3 can be absorbed on the catalyst surface physically and chemically. The physically absorbed NH3 is desorbed at lower temperatures, whereas the chemically absorbed NH3 is desorbed at higher temperatures. The low desorption temperature peak is assigned to the desorption of ammonia over weak acidic sites, and the high desorption temperature peak is allocated to the desorption of ammonia from strong acid sites, which is related to the Brönsted acid sites presented on the catalyst. Fig. 7 shows the NH3-TPD curves of the NiMo/Al2O3–AT-r-25, NiMo/Al2O3–AT-r-200, NiMo/Al2O3, NiMo/AT-r-25 and NiMo/AT-r-200 catalysts. Both NiMo/Al2O3–AT-r-25 and NiMo/Al2O3–AT-r-200 possess broad desorption peaks from 100 to 500 °C. Over these two samples, a broad peak is presented near 270 °C, NiMo/Al2O3–AT-r-25 exhibits a weak shoulder peak at 130 and 350 °C, which indicates the presence of a certain amount of weak, and medium acid sites in the aluminum-modified TUD-1 sample. As for NiMo/AT-r-25 and NiMo/AT-r-200, the desorption peaks range from 100 to 300 °C, which indicate that these two catalysts have less strong acid sites compared with the NiMo/Al2O3–AT-r-25 and NiMo/Al2O3–AT-r-200 catalysts. Furthermore, the released amount of NH3 increases with the decrease of the Si/Al ratio in the synthesis of AT-r. It is obvious that the incorporated aluminum amount in AT is regulated to the acidity of the catalysts. It is apparent to see that the support mixture of the Al2O3 and AT-r material has a stronger acidity than the separated AT-r materials, which indicates that Al2O3 in the support contributes more to the higher acidity of the catalyst compared to AT-r.
 | ||
| Fig. 7 NH3-TPD profiles of the NiMo/Al2O3–AT-r catalysts. (a) NiMo/Al2O3–AT-r-25; (b) NiMo/Al2O3–AT-r-200; (c) NiMo/Al2O3; (d) NiMo/AT-r-200; (e) NiMo/AT-r-25. | ||
The acidities of the supported catalysts were investigated by the pyridine-FTIR method, as shown in Fig. 8. Based on the literature,21,22 it is well known that the bands at 1540 and 1450 cm−1 are derived from the pyridine molecules strongly bound to Brönsted acid sites (B) and Lewis acid sites (L), respectively. The band at 1490 cm−1 is attributed to the adsorbed pyridine associated with both Lewis and Brönsted acid sites. The catalysts of NiMo/Al2O3, NiMo/Al2O3–AT-r-50 and NiMo/Al2O3–TUD-1 exhibit a strong and four weak absorbed bands due to the pyridine adsorption on Lewis acid sites (absorption peaks at 1450, 1576, 1610, 1622 and 1490 cm−1). No Brönsted acid sites are observed in the spectra of NiMo/Al2O3, as estimated from the absence of the absorption band of pyridine at 1540 cm−1; moreover, few Lewis acid sites are observed in the spectra of NiMo/AT-r-50 in Fig. 8A except for the weak intensity of the absorption band of pyridine at 1450 cm−1. In contrast, the bands at 1540 and 1635 cm−1 are found in the spectra of the NiMo/Al2O3–AT-r-50, and NiMo/Al2O3–TUD-1 catalysts, confirming the existence of Brönsted acid sites in these samples.
 | ||
| Fig. 8 The Py-IR profiles of NiMo/Al2O3–AT-r catalysts (A: 200 °C; B: 350 °C). (a) NiMo/Al2O3–TUD-1; (b) NiMo/Al2O3–AT-r-50; (c) NiMo/Al2O3; (d) NiMo/AT-r-50. | ||
The concentrations of Brönsted, Lewis, total acid sites and the ratios of B/L are given in Table 5. An increase in Brönsted acid sites concentration is clearly visible with increase in AT content. From Table 5, it is obvious that Al2O3 contributes no B acid sites to the related catalysts, whereas the amount of B acid sites in the NiMo/Al2O3–AT-r-50 catalyst is 62.8 μmol g−1, demonstrating that the AT-r material has an interaction with the Al2O3 carrier causing the increase of B acid sites in the catalyst. The temperature programmed reduction (H2-TPR) analysis was performed to determine the nature of the different Ni and Mo oxide species on the supports and the catalyst surface, as well as to study the interactions between NiMo and the supports in the calcined catalysts.
| Catalysts number | 200 °C | 350 °C | ||||||
|---|---|---|---|---|---|---|---|---|
| Acid content (μmol g−1) | Acid content (μmol g−1) | |||||||
| L | B | L + B | B/L | L | B | L + B | B/L | |
| NiMo/Al2O3 | 292.3 | 0 | 292.3 | 0 | 150.4 | 0 | 150.4 | 0 |
| NiMo/Al2O3–TUD-1 | 324.8 | 17.2 | 342.0 | 0.06 | 215.6 | 4.8 | 220.4 | 0.02 |
| NiMo/Al2O3–AT-r-50 | 299.8 | 62.8 | 362.6 | 0.21 | 184.7 | 8.9 | 193.6 | 0.05 |
| NiMo/AT-r-50 | 36.0 | 15.2 | 51.2 | 0.42 | 4.7 | 7.1 | 11.8 | 1.51 |
The H2-TPR characterizations of the NiMo series catalysts are shown in Fig. 9. The TPR profiles of the samples (Fig. 9a–c) show two main reduction peaks centered at 500–600 °C and ∼820 °C. The other samples exhibit two main reduction peaks centered at about 400–450 and 770–800 °C. The reduction peak located at about 420 °C can be assigned to the first step of the reduction Mo6+ → Mo4+ of which the amorphous, highly defective, multilayered octahedral Mo species are generally believed to be the precursors of the active type-II Ni–Mo–S phases.37 A weak shoulder peak observed at about 530 °C could be assigned to the reduction of the Ni2+ species or NiMoO4 on the Al2O3–AT-r surfaces.38 The reduction peak located at about 790 °C is ascribed to the deep reduction of all the Mo species that are intensely bound to the composite supports, including highly dispersed tetrahedral Mo species, intermediate-reducible crystalline phases of orthorhombic MoO3 or Al2(MoO4)3 and the second step of reduction of the polymeric octahedral Mo4+ → Mo0. Furthermore, no bulk MoO3 peak is observed over all samples in the temperature range of 600–630 °C, confirming that the Mo species have a good distribution on the support,39 which is in accordance with the XRD characterization results. With increase in aluminum content, the TPR peaks belonging to the first reduction step of Mo6+ → Mo4+ shift to a higher temperature, indicating that the incorporation of aluminum in the supports has great influence on the redox properties of the catalysts. The change in the reducibility of Mo with the aluminum contents correlates to the MSI between Mo and the support. More aluminum species incorporated into the support resulted in stronger MSI in the catalysts. The Mo species on the series catalysts (Fig. 9a–d) are more difficult to reduce, whereas the combination of Al2O3 and the AT-r materials makes the reduction for MoO3 on the supported catalysts more easier, which indicates that the supports containing Al2O3 and AT-r materials have an appropriate MSI compared to others.
 | ||
| Fig. 9 H2-TPR profiles of the NiMo/Al2O3–AT-r catalysts. (a) NiMo/AT-r-25; (b) NiMo/AT-r-50; (c) NiMo/AT-r-100; (d) NiMo/Al2O3; (e) NiMo/Al2O3–AT-25; (f) NiMo/Al2O3–AT-50; (g) NiMo/Al2O3–AT-100. | ||
3.3 HDS performances of the catalysts
In this research, all catalysts of NiMo/Al2O3–AT-r with different Si/Al ratios and NiMo/Al2O3–AT-m with different AT additions were prepared by using AT-r and AT-m as additives, respectively. In order to study the suitable addition of AT contents in the catalysts, the catalytic activities of the NiMo/Al2O3–AT-m catalysts were evaluated by the HDS efficiencies using PetroChina Hohhot petrochemical company FCC diesel as feedstock, and the results are listed in Table 6.| Catalyst | Mass contents of AT in support (%) | Total sulfur of hydrotreated oil (mg L−1) | HDS efficiency (%) |
|---|---|---|---|
| a AT was synthesized with Si/Al of 50 at 180 °C for 18 h. | |||
| NiMo/Al2O3 | 0 | 56.9 | 94.4 |
| NiMo/Al2O3–AT-m-10% | 10 | 31.7 | 96.9 |
| NiMo/Al2O3–AT-m-20% | 20 | 29.9 | 97.0 |
| NiMo/Al2O3–AT-m-30% | 30 | 26.2 | 97.4 |
| NiMo/Al2O3–AT-m-40% | 40 | 81.6 | 92.0 |
| NiMo/Al2O3–AT-m-50% | 50 | 124.7 | 87.7 |
| NiMo/Al2O3–AT-m-80% | 80 | 157.1 | 84.5 |
| NiMo/AT | 100 | 190.2 | 81.2 |
Obviously, the evaluation data proved that the best catalyst was NiMo/Al2O3–AT-m-30% with 30 m% AT in the support and it exhibited the highest HDS efficiency, as high as 97.4%, and the sulfur content of the product is 26.2 mg L−1.
Based on the fact that the most suitable AT content in the support is 30%, the effects of the AT synthetic parameters including Si/Al, heat treatment time and heat treatment temperature were systematically studied. Table 7 lists the HDS activities of the NiMo/Al2O3–AT-r series catalysts with different Si/Al ratios of the AT composite. The results show that the HDS efficiencies of the catalysts increase with the Si/Al ratios and reach a maximal value when the Si/Al ratio is 50 (S as low as 26.2 mg L−1), which is relatively higher than those catalysts of NiMo/Al2O3 and NiMo/Al2O3–TUD-1. The result may be explained by the suitable content of aluminum incorporation in the AT, which could improve the acidity of the catalyst, and then enhance the breaking ability of the C–S bond in sulfur compounds.40,41
| Catalyst | Si/Al ratio | S/mg L−1 | HDS/% |
|---|---|---|---|
| NiMo/Al2O3 | Al2O3 | 56.9 | 94.4 |
| NiMo/Al2O3–AT-r-25 | 25 | 50.3 | 95.0 |
| NiMo/Al2O3–AT-r-50 | 50 | 30.2 | 97.4 |
| NiMo/Al2O3–AT-r-75 | 75 | 36.2 | 96.4 |
| NiMo/Al2O3–AT-r-100 | 100 | 53.5 | 94.7 |
| NiMo/Al2O3–AT-r-200 | 200 | 66.7 | 93.4 |
| NiMo/Al2O3–TUD-1 | Si-TUD-1 | 63.9 | 93.7 |
Fig. 10 and 11 show the HDS and HDN activities of the sulfide NiMo/Al2O3–AT-ti catalysts and NiMo/Al2O3–AT-te using FCC diesel oil as the feedstock. As can be seen in Fig. 10 and 11, all catalysts exhibited high HDS and HDN conversions (>92%), especially HDN is greater than 95%. Along with increase in heat time and heat temperature, the HDS conversion reaches a maximum of 97.4% when the heat time is 18 h. Moreover, the HDS conversion has a peak value at the heat temperature of 180 °C. These catalytic results could be attributed to the texture properties of the materials such as specific surface area, pore diameter and total pore volume (in Tables 2 and 3),10 since suitable synthesis conditions are important to construct favorable frameworks of a mesoporous material with unrestricted pore channels.
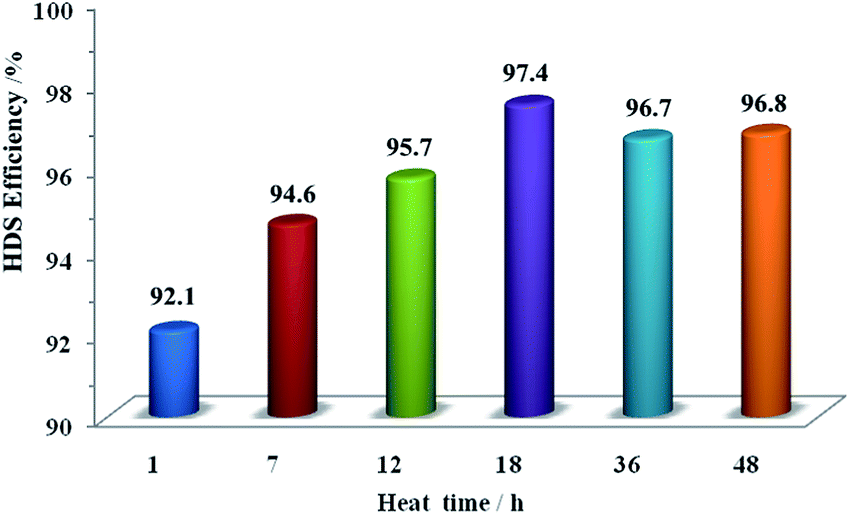 | ||
| Fig. 10 HDS activities of the sulfided NiMo/Al2O3–AT-ti catalysts. Note: all samples were synthesized with Si/Al = 50; heat temperature of 180 °C. | ||
 | ||
| Fig. 11 HDS (square column) and HDN (cylinder) activities of NiMo/Al2O3–AT-te catalysts. Note: all samples were synthesized with Si/Al = 50, heat time of 18 h. | ||
From the data in Fig. 11, it could be observed that the HDN efficiencies showed similar trends as the HDS results. In particular, all of the NiMo/Al2O3–AT-te series catalysts show high HDN efficiencies, as high as 99.6%, and the nitrogen content in products is as low as 2.4 mg L−1. Moreover, the HDN conversion is almost independent to the heat temperature.
Combining the catalytic performances of the above four series catalysts, it can be demonstrated distinctly that the catalysts incorporated with AT composites not only have good HDS activities but also high HDN efficiencies, and the NiMo/Al2O3–AT catalyst with an AT addition of 30 m%, Si/Al ratio of 50, heat time of 18 h and heat temperature of 180 °C achieves the highest HDS and HDN efficiencies, which are 97.4% for HDS and 99.6% for HDN. The main reasons might be interpreted by the following aspects: (i) aluminum-modified 3D mesoporous TUD-1 materials can reduce the MSI, and lead to the formation of octahedral Mo species, which are the precursors to form more active sites that enhance the hydrogenation activity of the catalysts;42 (ii) the unrestricted channels of the AT composites lessen the molecular diffusional limitation by improving the specific surface areas and total pore volumes of the supported catalysts; and (iii) the incorporation of the AT material into the catalysts enhances the hydrogenolysis ability, which may be attributed to the total amount of acid sites, especially for the weak-moderate intensity acid sites that play an important role in the hydrogenation process.8 Furthermore, the large amounts of strong acid leads to the easier cleavage of the C–N bond,43 which favors improvement in HDN efficiencies.44
4. Conclusions
In this research, aluminum-modified 3D mesoporous TUD-1 material was synthesized by a sol–gel method using TEA and TEAOH as the co-template. The characterization results of XRD and TEM proved that aluminum was incorporated into the framework of the TUD-1 silicate network, and the as-synthesized material had a uniform pore size distribution, high surface area and high pore volume. The corresponding supported NiMo catalysts were prepared by using AT materials as the support additives. The HDS performances of the series catalysts were evaluated using FCC diesel as the feed. In the series of NiMo/Al2O3–AT-m catalysts, the catalyst with 30 m% AT in the support exhibited the highest HDS conversion, which was as high as 97.42%. Moreover, the effects of the synthetic parameters, including the Si/Al ratio, heat treatment time and heat temperature were systematically studied, and the hydrotreating results showed that the catalyst with the Si/Al ratio of 50 in AT, heat time of 18 h and heat temperature of 180 °C exhibited the highest HDS (97.56%) and HDN (99.63%) conversions.Acknowledgements
The authors gratefully acknowledge the financial supports from National Natural Science Foundation of China (no. 21276277), Opening Project of Guangxi University KLPRPPIT (2012K01) and CNPC Key Research Project.Notes and references
- V. C. Srivastava, RSC Adv., 2012, 2, 759–783 RSC.
- M. M. Maricq, R. E. Chase, N. Xu and P. M. Laing, Environ. Sci. Technol., 2002, 36, 283–289 CrossRef CAS.
- Y. Shiraishi, K. Tachibana, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2002, 41, 4362–4375 CrossRef CAS.
- C. S. Song and X. L. Ma, Appl. Catal., B, 2003, 41, 207–238 CrossRef CAS.
- T. Fujikawa, H. Kimura, K. Kiriyama and K. Hagiwara, Catal. Today, 2006, 111, 188–193 CrossRef CAS PubMed.
- C. S. Song, Catal. Today, 2003, 86, 211–263 CrossRef CAS.
- A. Stanislaus, A. Marafi and M. S. Rana, Catal. Today, 2010, 153, 1–68 CrossRef CAS PubMed.
- D. Q. Zhang, A. J. Duan, Z. Zhao and C. M. Xu, J. Catal., 2010, 247, 273–286 CrossRef PubMed.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- J. C. Jansen, Z. Shan, L. Marchese, W. Zhou, N. v. d. Puil and T. Maschmeyer, Chem. Commun., 2001, 713–714 RSC.
- S. Telalović, A. Ramanathan and G. Mul, J. Mater. Chem., 2010, 20, 642–658 RSC.
- Z. Shan, J. C. Jansen, W. Zhou and T. Maschmeyer, Appl. Catal., A, 2003, 254, 339–343 CrossRef CAS.
- R. Anand, R. Maheswari and U. Hanefeld, J. Catal., 2006, 242, 82–91 CrossRef CAS PubMed.
- V. Valtchev, S. Mintova and M. Tsapatsis, Ordered Porous Solids: Recent Advances and Prospects, Elsevier, 2009, pp. 3–29 Search PubMed.
- Z. X. Zhang, P. Bai, B. J. Xu and Z. F. Yan, J. Porous Mater., 2006, 13, 245–250 CrossRef CAS PubMed.
- R. Bala, US Pat., 0 009 665, 2006.
- D. P. Liu, X. Y. Quek, S. Q. Hu, L. S. Li, H. M. Lim and Y. H. Yang, Catal. Today, 2009, 147, S51–S57 CrossRef CAS PubMed.
- Z. P. Shan, C. J. Jansen, C. Y. Yeh, P. J. Angevine and T. Maschmeyer, US Pat., 7 211 238, 2007.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- T. Barzetti, E. Selli, D. Moscotti and L. Forni, J. Chem. Soc., 1996, 92, 1401–1407 CAS.
- M. S. Hamdy, J. Mol. Catal. A: Chem., 2014, 393, 39–46 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- J. C. Groen, L. A. A. Peffer and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS.
- S. Telalović, S. K. Karmee, A. Ramanathan and U. Hanefeld, J. Mol. Catal. A: Chem., 2013, 368–369, 88–94 CrossRef PubMed.
- P. Neves, M. M. Antunes, P. A. Russo, J. P. Abrantes, S. Lima, A. Fernandes, M. Pillinger, S. M. Rocha, M. F. Ribeiro and A. A. Valente, Green Chem., 2013, 15, 3367–3376 RSC.
- S. Lima, M. M. Antunes, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal. A, 2010, 388, 141–148 CrossRef CAS PubMed.
- D. Q. Zhang, A. J. Duan, Z. Zhao and C. M. Xu, J. Catal., 2010, 274, 273–286 CrossRef CAS PubMed.
- Y. Han, F. S. Xiao, S. Wu, Y. Y. Sun, X. J. Meng, D. S. Li and S. Lin, J. Phys. Chem. B, 2001, 105, 7963–7966 CrossRef CAS.
- C. Simons, U. Hanefeld, I. W. C. E. Arends, R. A. Sheldon and T. Maschmeyer, Chem.–Eur. J., 2004, 10, 5829–5835 CrossRef CAS PubMed.
- R. Prins, G. Ertl, H. Knözinger and J. Weitkam, Handbook of heterogeneous catalysis, Wiley–VCH, Weinheim, 1997, p. 1908 Search PubMed.
- C. C. Williams, J. G. Ekerdt, J. M. Jehng, F. D. Hardcastle, A. M. Turek and I. E. Wachs, J. Phys. Chem., 1991, 95, 8781–8791 CrossRef CAS.
- Z. Liu and Y. Chen, J. Catal., 1998, 177, 314–324 CrossRef CAS.
- R. S. Weber, J. Catal., 1995, 151, 470–474 CrossRef CAS.
- P. Salerno, S. Mendioroz and A. A. López, Appl. Catal., A, 2004, 259, 17–28 CrossRef CAS PubMed.
- H. Topsøe, B. S. Clausen, R. Candia, C. Wivel and S. Mørup, J. Catal., 1981, 68, 433–452 CrossRef.
- S. Chaturvedi, J. A. Rodriguez and J. L. Brito, Catal. Lett., 1998, 51, 85–93 CrossRef CAS.
- T. Ressler, J. Wienold and R. E. Jentoft, Solid State Ionics, 2001, 141–142, 243–251 CrossRef CAS.
- Q. Huo, T. Dou, Z. Zhao and H. F. Pan, Appl. Catal., A, 2010, 381, 101–108 CrossRef CAS PubMed.
- O. Y. Gutiérrez, D. Valencia, G. A. Fuentes and T. Klimova, J. Catal., 2007, 249, 140–153 CrossRef PubMed.
- D. Solís, T. Klimova, R. Cuevas, J. Ramírez and A. López-Agudo, Catal. Today, 2004, 98, 201–206 CrossRef PubMed.
- F. Rota and R. Prins, J. Mol. Catal. A: Chem., 2000, 162, 367–374 CrossRef.
- R. Prins, Adv. Catal., 2001, 46, 399–464 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra10777b |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |