
Simple synthesis of bimetallic alloyed Pd–Au nanochain networks supported on reduced graphene oxide for enhanced oxygen reduction reaction†
Qian-Li Zhangab,
Jin-Xia Fengb,
Ai-Jun Wanga,
Jie Weia and
Jiu-Ju Feng*a
aCollege of Chemistry and Life Science, College of Geography and Environmental Science, Zhejiang Normal University, Jinhua 321004, China. E-mail: jjfeng@zjnu.cn; Fax: +86 579 82282269; Tel: +86 579 82282269
bSchool of Chemistry and Biological Engineering, Suzhou University of Science and Technology, Suzhou, 215009, China
First published on 14th October 2014
Abstract
Bimetallic alloyed Pd–Au nanochain networks supported on reduced graphene oxide (Pd–Au NNs/RGO) were prepared by a one-pot wet-chemical co-reduction method with the assistance of caffeine as a capping agent and a structure directing agent, while no seed, template, or surfactant was involved. It was found that the dosage of caffeine and the concentrations of the precursors (i.e. PdCl42− + AuCl4−) played essential roles in the formation of Pd–Au NNs. Moreover, the as-prepared nanocomposites exhibited much better electrocatalytic performance than those of conventional Pd black and Pd–C toward oxygen reduction reaction (ORR) in alkaline media in terms of the onset potential, limiting current, and stability.
1. Introduction
Pt and Pt-based catalysts are generally considered as the most efficient electrocatalysts for oxygen reduction reaction (ORR) in fuel cells, owing to their superior catalytic activity toward direct reduction of oxygen to water via the 4-electron pathway.1 Unfortunately, these catalysts are seriously inhibited from large-scale commercial applications because of their high cost, limited supply, poor stability, and sluggish kinetics for ORR.2,3 Therefore, it is urgent to explore non-Pt catalysts to reduce costs and improve the catalytic activity during ORR. Several good examples are reported in the past, such as Pd,4 Ir,5 and Ru-based6 bimetallic catalysts, oxides,7,8 carbides,9 and non-precious catalysts.10–13Among various non-Pt catalysts, Pd-based catalysts have attracted great interest as cathode materials in fuel cells, owing to their relatively low cost and more abundance as compared to Pt-based catalysts.14–17 Particularly, the incorporation of a second metal (M) into Pd (PdM) often brings better catalytic performance, compared to individual counterparts, owing to the synergistic effects and rich diversity of the compositions.18,19
Lately, several PdM catalysts demonstrate the enhanced catalytic activity for ORR, including Pd–Au,20 Pd–Ni,21 Pd–Co,22 Pd–Mo,23 Pd–Ti,24 Pd–Ag,25 and Pd–Cu26 catalysts. Among them, Pd–Au catalysts have attracted increasing attention for their striking catalytic activity toward ORR.27–29 For example, Son et al. synthesized sponge-like Pd/Au with higher catalytic activity than single Pd with the similar morphology, commercial Pd-20/C, and Pt-20/C catalysts for ORR.30 Koenigsmann and coworkers fabricated Pd9Au nanowires with superior catalytic performance for oxygen reduction, compared with commercial Pt nanoparticles.31
In spite of their enhanced catalytic performances toward ORR, their synthesis is complex, which are usually involved toxic organic solvent, high temperature, time-consuming, sacrificial templates, and complicated synthetic procedures. For instance, Shim and coworkers prepared carbon-supported Pd-coated Au nanoparticles by spontaneous reduction of Pd(II) to Pd on a Au nanoparticles surface, with the reaction time of about ten hours.20 In another example, Cha's group synthesized carbon-supported porous Pd shell coated Au NNs, using Co nanoframes as sacrificial templates.28 Therefore, it is of great significance to develop a common, rapid, and environmental friendly approach to fabricate Pd–Au catalysts with the improved electrocatalytic performance.
Given that the catalytic activity of Pd–Au catalysts is not only affected by the morphology, size and dispersion, but also by the nature of a support that is associated with the distribution of the catalysts.32,33 Graphene oxide (GO) has abundant oxygen-containing functional groups on the surface (e.g. –OH, –COOH, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), which can be used as anchoring sites for metallic catalysts, along with contributing to its large surface area.34,35 Hence, the presence of GO or reduced GO (RGO) is benefit to the catalytic activity of the as-prepared nanocomposites for ORR, as verified by previous work.36,37
O), which can be used as anchoring sites for metallic catalysts, along with contributing to its large surface area.34,35 Hence, the presence of GO or reduced GO (RGO) is benefit to the catalytic activity of the as-prepared nanocomposites for ORR, as verified by previous work.36,37
Herein, a simple, rapid, and one-pot wet-chemical co-reduction method was explored for synthesis of alloyed Pd–Au nanochains networks uniformly anchored on reduced graphene oxide (denoted as Pd–Au NNs/RGO), with the assistance of caffeine. The electrocatalytic performance of Pd–Au NNs/RGO was investigated, using ORR as a model system.
2. Experimental
2.1 Chemicals
Graphite powder (99.95%, 8000 mesh), HAuCl4, PdCl2, hydrazine hydrate (80%), caffeine, commercial Pd black and 10% Pd–C were purchased from Shanghai Aladdin Chemical Reagent Company (Shanghai, China). All the other chemicals were of analytical grade and used as received. 1.77 g PdCl2 was dissolved with 0.4 mL HCl (37%), and then diluted to 100 mL with water (100 mM H2PdCl4). All solutions were prepared with twice-distilled water throughout the whole experiment.2.2 Synthesis of Pd–Au NNs/RGO
For typical synthesis of Pd–Au NNs/RGO, GO was firstly prepared from purified natural graphite by a modified Hummer's method.38 Next, 0.5 mL GO (0.5 mg mL−1) was mixed with 10 mL of aqueous caffeine solution (25 mM) in the ice bath (0 °C), followed by gradual addition of 0.5 mL of 100 mM H2PdCl4 and 0.93 mL of 53.95 mM HAuCl4 under stirring. Afterward, 0.2 mL of hydrazine hydrate (80%) was slowly dropped into the mixed solution. The solution color quickly changed from yellow to black, which was further reacted for 15 min. The black precipitate was washed and recollected by consecutive washing/centrifugation cycles with water, and dried at 60 °C in a vacuum oven for further characterization.2.3 Characterization
The morphologies and sizes of the samples were characterized by transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) on a JEM-2010HR transmission electron microscope operated at 200 kV equipped with an energy-dispersive X-ray spectrometer (EDS). The samples were prepared by ultrasonic dispersion in ethanol and depositing a drop of the suspension on a copper grid. The crystalline structures were determined by X-ray diffraction (XRD) spectroscopy, which were performed on a Bruker-D8-AXS diffractometer system equipped with Cu Kα radiation (Bruker Co., Germany). The elemental composition analysis was measured by X-ray photoelectron spectroscopy (XPS), which were conducted by a K-Alpha XPS spectrometer (ThermoFisher, E. Grinstead, UK), using Al Kα X-ray radiation (hν = 1486.6 eV). Raman spectra were recorded on a Renishaw Raman system model 1000 spectrometer equipped with a charge coupled device (CCD) detector. Fourier-transform infrared (FT-IR) spectra were recorded on a Nicolet NEXUS-670 FT-IR spectrometer from 500 to 4000 cm−1. Thermogravimetric analysis (TGA, NETZSCH STA 449C) was performed with a heating rate of 10 °C min−1 in air atmosphere from room temperature to 800 °C.2.4 Electrochemical measurements
Electrochemical measurements were carried out on a CHI 660D electrochemical workstation (CH Instruments, Chenhua Co., Shanghai, China). A conventional three electrode cell was used, including an Ag–AgCl electrode as reference electrode, a platinum wire as counter electrode, and a bare, modified glassy carbon electrode (GCE, 3 mm in diameter) or rotating disk electrode (RDE, 4 mm in diameter) as working electrode.For fabrication of Pd–Au NNs/RGO modified electrode, 4 mg of Pd–Au NNs/RGO was dispersed in 2 mL water under ultrasonication for 30 min to obtain a homogenous suspension. Then, 8 and 20 μL of the resulting suspension were deposited on the pre-cleaned GCE and RDE, respectively, and allowed to dry in air. After drying, 4 and 10 μL of Nafion (0.05 wt%) were deposited onto the electrode surfaces, respectively. For comparison, Pd-black and Pd–C modified electrodes were prepared in a similar way.
The electrochemically active surface area (ECSA) of the catalysts modified GCE was calculated by the CO stripping experiments. CO gas was firstly adsorbed on the modified GCE at 0.1 V for 900 s in 0.5 M CO-saturated H2SO4, and then recorded the corresponding CO-stripping voltammograms in H2SO4.
The electrocatalytic performances of the catalysts modified RDE were determined in 0.1 M oxygen-saturated KOH at a scan rate of 5 mV s−1 from 100 to 3600 rpm. Meanwhile, the stability tests were conducted in oxygen-saturated 0.1 M KOH at the half-wave potential by chronoamperometry, with a rotating rate of 1600 rpm. All the tests were performed at room temperature, if not sated otherwise.
3. Results and discussion
3.1 Characterization of Pd–Au NNs/RGO
The morphology and size of Pd–Au NNs/RGO were examined by TEM analysis (Fig. 1). As can be seen, the product contains a lot of typical Pd–Au NNs well dispersed on RGO surface (Fig. 1A and B). The absence of caffeine yields heavy aggregated nanoparticles (Fig. S1A, ESI†). Alternatively, excessive caffeine induces the destruction of the network-like structures (e.g. 50 mM, Fig. S1B†). Therefore, caffeine is critical for synthesis of Pd–Au NNs, which serves as a capping agent and a structure directing agent to effectively prevent the aggregation of Pd–Au nanocrystals on RGO sheets.39 | ||
| Fig. 1 Representative low- (A and B) and high- (C and D) resolution TEM images of Pd–Au NNs/RGO. | ||
Besides, less (e.g. 1 mM) and excess (e.g. 7.5 mM) amounts of the total precursors (i.e. PdCl42− and AuCl4−) produce seriously aggregated Pd–Au nanoparticles (Fig. S2, ESI†). These observations imply the essential role of the precursors with appropriate concentrations in the present system.
HRTEM images show good crystallinity of Pd–Au NNs (Fig. 1C and D), as indicated by their well-defined fringe pattern. Moreover, the inter-planar distance between two adjacent fringes is about 0.233 nm, corresponding to the (111) planes of face-centered cubic (fcc) Pd–Au alloy.40 These observations indicate that the precursors are simultaneously reduced by hydrazine hydrate to form alloy at the very early stage, and Pd–Au nanocrystals grow along the (111) directions.41,42
Meanwhile, HAADF-STEM-EDS mapping and line scanning profiles were conducted to examine the elemental distributions in Pd–Au NNs. As displayed in Fig. 2, Au and Pd atoms are uniformly distributed across the whole nanochains networks. It means the formation of Pd–Au alloy, rather than the mixture of Pd and Au.
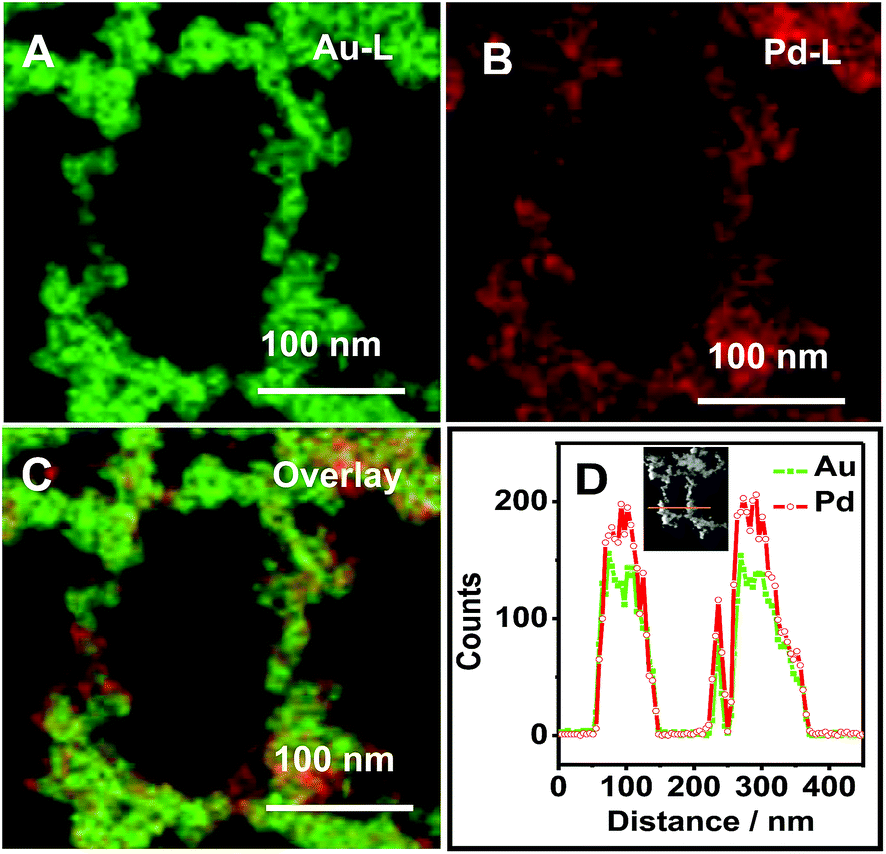 | ||
| Fig. 2 HAADF-STEM-EDS mapping (A–C) and EDS line scanning profiles (D) of an individual Pd–Au NN. | ||
EDS (Fig. 3A) and XRD (Fig. 3B) measurements were carried out to evaluate the composition and crystal structures of Pd–Au NNs/RGO. EDS analysis shows that the content of Au is more abundant than that of Pd in Pd–Au NNs, with Pd to Au at atomic ratio of 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.8. The difference is attributed to different reduction potentials of Au3+ (AuCl4−/Au, 1.002 V vs. SHE) and Pd2+ (PdCl42−/Pd, 0.591 V vs. SHE). Notably, Au3+ is more easily reduced, compared with Pd2+, leading to the formation of Au-rich alloys.
:1.8. The difference is attributed to different reduction potentials of Au3+ (AuCl4−/Au, 1.002 V vs. SHE) and Pd2+ (PdCl42−/Pd, 0.591 V vs. SHE). Notably, Au3+ is more easily reduced, compared with Pd2+, leading to the formation of Au-rich alloys.
 | ||
| Fig. 3 (A) EDS pattern of Pd–Au NNs/RGO. (B) XRD patterns of Pd–Au NNs/RGO (curve a), GO (curve b), and the standard patterns of bulk Pd and Au. | ||
Fig. 3B shows the XRD spectrum of Pd–Au NNs/RGO (curve a), in which the representative peak at 38.4° emerges between the diffraction peaks of the Au (111) and Pd (111) crystal planes, confirming the formation of Pd–Au alloy.43 And a broad peak at 21.2° is assigned to the (002) planes of RGO,44 which is different from that of GO with a sharp peak at 11.0° (Fig. 3B, curve b), indicating the effective reduction of GO.45
XPS is an effective method to investigate elemental composition and valence state of materials. Fig. 4A shows that the peaks located at 532.6, 335.3, 284.5, and 85.1 eV correspond to O 1s, Pd 3d, C 1s, and Au 4f in the survey XPS spectrum, respectively, revealing the presence of Pd and Au in Pd–Au NNs/RGO. In the high-resolution Pd 3d region (Fig. 4B), the binding energies centered at 341.1 eV (Pd 3d3/2) and 335.6 eV (Pd 3d5/2), 341.9 eV (Pd 3d3/2) and 336.5 eV (Pd 3d5/2) are assigned to metallic Pd0 and Pd2+,46 respectively. While only one pair of peaks is emerged at 87.8 and 84.1 eV in the Au 4f region (Fig. 4C), which is ascribed to 4f5/2 and 4f7/2 of metallic Au0,47 revealing complete reduction of AuCl4− in the present synthesis. All these results further confirm that metallic Pd0 and Au0 are the predominant species.
 | ||
| Fig. 4 Survey (A), and high-resolution Pd 3d (B), Au 4f (C), and C 1s (D) XPS spectra of Pd–Au NNs/RGO. | ||
For the C 1s XPS spectrum (Fig. 4D), there is a strong peak at 248.7 eV corresponding to the C–C bonds, along with three relatively weaker ones at 286.5, 287.5, and 288.8 eV associated with the C–O, CO, and O–CO bonds, respectively.48,49 It indicates that GO is reduced to RGO, as strongly supported by the FT-IR and Raman analysis.
As depicted in Fig. 5A, the FT-IR spectrum of Pd–Au NNs/RGO (Fig. 5A, curve a) clearly shows that the characteristic peak of CO stretching vibration of carboxyl at 1733.5 cm−1 and C–O stretching vibration of epoxy and alkoxy at 1058.2 cm−1 are weaker, compared with the CC skeletal vibration at 1663.2 cm−1,50 using the spectrum of GO (Fig. 5A, curve b) as a standard. It reveals the efficient removal of most oxygen containing functional groups.
 | ||
| Fig. 5 FT-IR (A) and Raman (B) spectra of Pd–Au NNs/RGO (curve a) and GO (curve b). | ||
Meanwhile, as illustrated by Raman spectra (Fig. 5B), the peak intensity ratio of D band (the E2g mode of sp2 carbon atoms) to G band (the A1g breathing mode of disordered graphite structure) for Pd–Au NNs/RGO (Fig. 5B, curve a) increases significantly, using the Raman spectrum of GO as a reference (Fig. 5B, curve b). This change suggests the formation of smaller graphene sp2 domains after the reduction of GO48 and at the same time confirms the effective reduction of GO to RGO again.
The thermal stability of Pd–Au NNs/RGO was examined by TGA (Fig. S3, curve a, ESI†). It is worth noting that there is a slight weight loss below 100 °C, which is attributed to the loss of absorbed water molecules on the surface, no obvious weight loss is detected when the sample is further heated up to 800 °C. This phenomenon is quite different from that of GO (Fig. S3, curve b, ESI†) under the identical conditions, in which GO shows two distinguished drops at 200 and 500 °C corresponding to the decomposition of oxygen-containing functional groups and carbon oxidation,51 respectively. These results combined with the above analysis further demonstrate the efficient reduction of GO to RGO. Additionally, the load of Pd–Au NNs is about 91 wt% in Pd–Au NNs/RGO.
3.2 Electrochemical measurements
Cyclic voltammetry is a convenient and efficient method to determine the electrochemical activity of a catalyst on an electrode. Fig. S4 (ESI†) provides the typical cyclic voltammograms (CVs) of Pd–Au NNs/RGO (curve a) and Pd-black (curve b) modified electrodes in 0.5 M H2SO4. It can be seen that hydrogen adsorption/desorption charges (QHads/QHdes) of Pd–Au NNs/RGO are higher than those of Pd-black, revealing the enlarged ECSA and more active sites available for Pd–Au NNs/RGO toward ORR.52As known, bulk Pd can absorb hydrogen to form Pd hydride,53 and hence the ECSA of catalysts were measured by CO stripping experiments. Fig. 6 offers the ECSA value of the catalysts, which are obtained by eqn (1):54
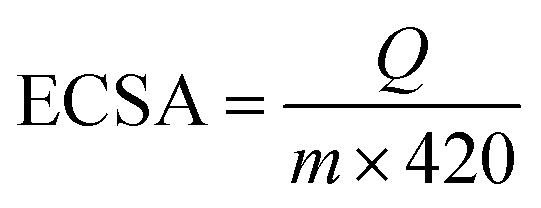 | (1) |
 | ||
| Fig. 6 CO-stripping voltammograms of Pd–Au NNs/RGO (A) and Pd black (B) modified GCE in 0.5 M H2SO4 at a scan rate of 50 mV s−1. | ||
To examine the electrocatalytic activity of Pd–Au NNs/RGO, the ORR corresponding polarization curves were recorded in oxygen-saturated 0.1 M KOH, using Pd-black and Pd–C as references. As displayed in Fig. 7A, the onset potential and half-wave potential (E1/2) for Pd–Au NNs/RGO (Fig. 7A, curve a) are −0.03 V and −0.13 V toward O2 reduction, respectively, which are much more positive than those of Pd-black (Fig. 7A, curve b) and Pd–C (Fig. 7A, curve c), highlighting that Pd-Au NNs facilitate ORR more favorably.55 Therefore, Pd–Au NNs/RGO modified electrode exhibits much higher diffusion-limiting current density than those of Pd-black and Pd–C. Importantly, the mass activity of Pd–Au NNs/RGO at −0.1 V is found to be the highest among the three catalysts (Fig. 7B). It means that Pd–Au NNs/RGO with the significantly improved catalytic activity toward oxygen reduction.
 | ||
| Fig. 7 (A) LSVs of Pd–Au NNs/RGO (curve a), Pd black (curve b), Pd–C (curve c), modified RDE in oxygen-saturated 0.1 M KOH at a scan rate of 5 mV s−1, using the rotation rate of 1600 rpm. (B) The corresponding mass activity at −0.1 V. (C) LSVs of Pd–Au NNs/RGO modified RDE with different rotating rates in oxygen-saturated 0.1 M KOH at a scan rate of 5 mV s−1 (curve a–f): 100, 400, 900, 1600, 2500, and 3600 rpm. (D) The corresponding K–L plots at different potentials for ORR. | ||
In order to examine the kinetics of ORR catalyzed by Pd–Au NNs/RGO, a series of polarization curves on Pd–Au NNs/RGO modified electrode were recorded in oxygen-saturated 0.1 M KOH with different rotation rates from 100 to 3600 rpm (Fig. 7C). Evidently, the limiting current densities increase with rotation rates, owing to the enhanced oxygen diffusion.56 Furthermore, the number of transferred electrons (n) for Pd–Au NNs/RGO is calculated by the below Koutechy–Levich equation:57,58
 | (2) |
| B = 0.2nFC0D2/30ν−1/6 | (3) |
500 C mol−1), C0 is the oxygen solubility (1.1 × 10−3 mol L−1), D0 is the diffusion coefficient of oxygen in 0.1 M KOH (1.9 × 10−5 cm2 s−1), and ν is the kinetic viscosity of the electrolyte (0.01 cm2 s−1).
As shown in Fig. 7D, the K–L plots (j−1 vs. ω−1/2) for Pd–Au NNs/RGO modified electrode at the potential ranging from −0.15 to −0.25 V exhibit good linearity, suggesting the first-order kinetics relative to the reactant concentration. And the electron transfer number is calculated from the slopes of K–L plots, with the values of 3.91, 3.82, and 3.90 electrons transferred at −0.15 V, −0.2 V, and −0.25 V, respectively, indicating approximate four-electron transfer pathway for Pd–Au NNs/RGO. That is, oxygen is directly reduced to water. The above discussion demonstrates that Pd–Au NNs/RGO is a compelling catalyst for oxygen reduction.
Fig. 8 shows the ORR polarization curves recorded by 1000 times in O2 saturated 0.1 M KOH on Pd–Au NNs/RGO modified electrode. After the durability test, similar polarization curve was observed in terms of the onset potential, half-wave potential and limiting current, evidencing the improved stability of Pd–Au NNs/RGO. It demonstrates Pd–Au NNs/RGO with improved stability toward ORR. On the other hand, the stability/durability of Pd–Au NNs/RGO modified electrode (Fig. 9, curve a) was also evaluated by chronoamperometry, using Pd-black (Fig. 9, curve b) and Pd–C (Fig. 9, curve c) as references. As illustrated in Fig. 9, the corresponding cathode current densities primarily show a gradual decay and then achieve a relatively steady state. Notably, Pd–Au NNs/RGO modified electrode (Fig. 9, curve a) exhibits considerably higher current and superior stability during the entire process, compared with Pd-black (Fig. 9, curve b) and Pd–C (Fig. 9, curve c). These results demonstrate that Pd–Au NNs/RGO is a stable electrocatalyst for ORR.
 | ||
| Fig. 8 The ORR polarization curves before and after the accelerated durability test at a scan rate of 5 mV s−1, using a rotation rate of 1600 rpm. | ||
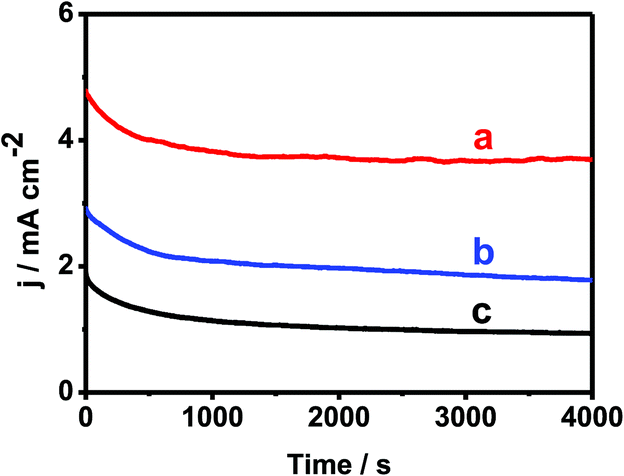 | ||
| Fig. 9 Chronoamperometric stability test of Pd–Au NNs/RGO (curve a), Pd black (curve b), and Pd–C (curve c) modified electrodes with a rotating rate of 1600 rpm in oxygen-saturated 0.1 M KOH at the half-wave potential. | ||
Taking the results from both activity and stability studies together, Pd–Au NNs/RGO has the enhanced electrocatalytic performances. This is ascribed to the following features: (I) promotional effects of Au in Pd–Au alloys,59 (II) synergistic effects between Au and Pd atoms,60,61 (III) well-dispersed Pd–Au NNs on RGO with high loading, (IV) unique structures of Pd–Au NNs make the catalyst better stability,28 and (V) graphene as a support in tuning electrocatalysis for highly efficient ORR.62
4. Conclusion
Well-defined bimetallic alloyed Pd–Au NNs were uniformly supported on RGO through one-pot wet-chemical co-reduction method, with the assistance of caffeine as a capping agent and a structure directing agent, which was simple, facile, and straightforward. Pd–Au NNs/RGO modified electrode exhibited much better electrochemical performance than commercial Pd-black and Pd–C toward ORR in terms of the onset potential, limiting current, and stability. The prepared Pd–Au NNs/RGO was a very promising cathodic catalyst in fuel cells. The synthetic method described herein can be extended to the preparation of other advanced RGO supported bimetallic catalysts.Acknowledgements
This work was financially supported by the National Natural Science foundation of China (21475118, 21175218, 21275130, and 51178283), Zhejiang province university young academic leaders of academic climbing project (pd2013055).References
- Y. Liu, Y.-Y. Wu, G.-J. Lv, T. Pu, X.-Q. He and L.-L. Cui, Electrochim. Acta, 2013, 112, 269–278 CrossRef CAS.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- G. Fu, K. Wu, J. Lin, Y. Tang, Y. Chen, Y. Zhou and T. Lu, J. Phys. Chem. C, 2013, 117, 9826–9834 CAS.
- K. Jukk, N. Alexeyeva, P. Ritslaid, J. Kozlova, V. Sammelselg and K. Tammeveski, Electrocatalysis, 2013, 4, 42–48 CrossRef CAS.
- J. Qiao, R. Lin, B. Li, J. Ma and J. Liu, Electrochim. Acta, 2010, 55, 8490–8497 CrossRef CAS.
- V. I. Zaikovskii, K. S. Nagabhushana, V. V. Kriventsov, K. N. Loponov, S. V. Cherepanova, R. I. Kvon, H. Bönnemann, D. I. Kochubey and E. R. Savinova, J. Phys. Chem. B, 2006, 110, 6881–6890 CrossRef CAS PubMed.
- K. Suito, A. Ishihara, M. Arao, M. Matsumoto, H. Imai, Y. Kohno, K. Matsuzawa, S. Mitsushima and K.-I. Ota, ECS Trans., 2013, 50, 1777–1783 CrossRef.
- N. Uehara, A. Ishihara, Y. Kohno, K. Matsuzawa, S. Mitsushima and K.-I. Ota, ECS Trans., 2013, 58, 1217–1223 CrossRef.
- K. Lee, A. Ishihara, S. Mitsushima, N. Kamiya and K.-I. Ota, Electrochim. Acta, 2004, 49, 3479–3485 CrossRef CAS.
- Y. Zhang, K. Fugane, T. Mori, L. Niu and J. Ye, J. Mater. Chem., 2012, 22, 6575–6580 RSC.
- N. Brun, S. A. Wohlgemuth, P. Osiceanu and M. M. Titirici, Green Chem., 2013, 15, 2514–2524 RSC.
- Z. Schnepp, Y. Zhang, M. J. Hollamby, B. R. Pauw, M. Tanaka, Y. Matsushita and Y. Sakka, J. Mater. Chem. A, 2013, 1, 13576–13581 CAS.
- S. Yasuda, L. Yu, J. Kim and K. Murakoshi, Chem. Commun., 2013, 49, 9627–9629 RSC.
- S. Takenaka, N. Susuki, H. Miyamoto, E. Tanabe, H. Matsune and M. Kishida, Chem. Commun., 2010, 46, 8950–8952 RSC.
- J. L. Fernández, V. Raghuveer, A. Manthiram and A. J. Bard, J. Am. Chem. Soc., 2005, 127, 13100–13101 CrossRef PubMed.
- M.-H. Shao, K. Sasaki and R. R. Adzic, J. Am. Chem. Soc., 2006, 128, 3526–3527 CrossRef CAS PubMed.
- O. Savadogo, K. Lee, K. Oishi, S. Mitsushima, N. Kamiya and K. I. Ota, Electrochem. Commun., 2004, 6, 105–109 CrossRef CAS.
- Y. Zhai, J. Zhai and S. Dong, Chem. Commun., 2010, 46, 1500–1502 RSC.
- J. Bao, W. Chen, T. Liu, Y. Zhu, P. Jin, L. Wang, J. Liu, Y. Wei and Y. Li, ACS Nano, 2007, 1, 293–298 CrossRef CAS PubMed.
- J. H. Shim, J. Kim, C. Lee and Y. Lee, Chem. Mater., 2011, 23, 4694–4700 CrossRef CAS.
- C. Xu, Y. Liu, Q. Hao and H. Duan, J. Mater. Chem. A, 2013, 1, 13542–13548 CAS.
- R. Rahul, R. Singh and M. Neergat, J. Electroanal. Chem., 2014, 712, 223–229 CrossRef CAS.
- C. J. Cao, X. G. Liu and Q. Liu, Adv. Mater. Res., 2013, 785, 390–394 CrossRef.
- Y. Liu and C. Xu, ChemSusChem, 2013, 6, 78–84 CrossRef CAS PubMed.
- C.-L. Lee, H.-P. Chiou, C.-M. Syu, C.-R. Liu, C.-C. Yang and C.-C. Syu, Int. J. Hydrogen Energy, 2011, 36, 12706–12714 CrossRef CAS.
- Y. Sha, T. H. Yu, B. V. Merinov and W. A. Goddard, ACS Catal., 2014, 4, 1189–1197 CrossRef CAS.
- J. S. Jirkovský, I. Panas, S. Romani, E. Ahlberg and D. J. Schiffrin, J. Phys. Chem. Lett., 2012, 3, 315–321 CrossRef.
- A. Cha, J. H. Shim, A. Jo, S. C. Lee, Y. Lee and C. Lee, Electroanalysis, 2014, 26, 723–731 CrossRef CAS.
- J.-N. Zheng, S.-S. Li, X. Ma, F.-Y. Chen, A.-J. Wang, J.-R. Chen and J.-J. Feng, J. Power Sources, 2014, 262, 270–278 CrossRef CAS.
- J. Son, S. Cho, C. Lee, Y. Lee and J. H. Shim, Langmuir, 2014, 30, 3579–3588 CrossRef CAS PubMed.
- C. Koenigsmann, E. Sutter, T. A. Chiesa, R. R. Adzic and S. S. Wong, Nano Lett., 2012, 12, 2013–2020 CrossRef CAS PubMed.
- T. Maiyalagan and K. Scott, J. Power Sources, 2010, 195, 5246–5251 CrossRef CAS.
- T. Maiyalagan, X. Wang and A. Manthiram, RSC Adv., 2014, 4, 4028–4033 RSC.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- S. I. Shin, A. Go, I. Y. Kim, J. M. Lee, Y. Lee and S.-J. Hwang, Energy Environ. Sci., 2013, 6, 608–617 CAS.
- Q.-L. Zhang, T.-Q. Xu, J. Wei, J.-R. Chen, A.-J. Wang and J.-J. Feng, Electrochim. Acta, 2013, 112, 127–132 CrossRef CAS.
- S. Xu, L. Yong and P. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 654–662 CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- H.-J. Choi, S.-M. Jung, J.-M. Seo, D. W. Chang, L. Dai and J.-B. Baek, Nano Energy, 2012, 1, 534–551 CrossRef CAS.
- S. C. Y. Tsen, P. A. Crozier and J. Liu, Ultramicroscopy, 2003, 98, 63–72 CrossRef CAS PubMed.
- F. Lan, D. Wang, S. Lu, J. Zhang, D. Liang, S. Peng, Y. Liu and Y. Xiang, J. Mater. Chem. A, 2013, 1, 1548–1552 CAS.
- X. Yu, D. Wang, Q. Peng and Y. Li, Chem.–Eur. J., 2013, 19, 233–239 CrossRef CAS PubMed.
- X. Gu, Z.-H. Lu, H.-L. Jiang, T. Akita and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11822–11825 CrossRef CAS PubMed.
- T.-Q. Xu, Q.-L. Zhang, J.-N. Zheng, Z.-Y. Lv, J. Wei, A.-J. Wang and J.-J. Feng, Electrochim. Acta, 2014, 115, 109–115 CrossRef CAS.
- S. Pei, J. Zhao, J. Du, W. Ren and H.-M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- Z. Jin, D. Nackashi, W. Lu, C. Kittrell and J. M. Tour, Chem. Mater., 2010, 22, 5695–5699 CrossRef CAS.
- H.-G. Boyen, G. Kästle, F. Weigl, B. Koslowski, C. Dietrich, P. Ziemann, J. Spatz, S. Riethmüller, C. Hartmann and M. Möller, Science, 2002, 297, 1533–1536 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- W. Tang, S. Jayaraman, T. F. Jaramillo, G. D. Stucky and E. W. McFarland, J. Phys. Chem. C, 2009, 113, 5014–5024 CAS.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- C. Xu, X. Wang, J. Zhu, X. Yang and L. Lu, J. Mater. Chem., 2008, 18, 5625–5629 RSC.
- X. Xue, C. Bock, L. Birry and B. MacDougall, Fuel Cells, 2011, 11, 286–300 CrossRef CAS.
- X.-M. Wang and Y.-Y. Xia, Electrochim. Acta, 2009, 54, 7525–7530 CrossRef CAS.
- Z.-B. Wang, G.-P. Yin, J. Zhang, Y.-C. Sun and P.-F. Shi, J. Power Sources, 2006, 160, 37–43 CrossRef CAS.
- Y.-B. Cho, J. E. Kim, J. H. Shim, C. Lee and Y. Lee, Phys. Chem. Chem. Phys., 2013, 15, 11461–11467 RSC.
- Z. Jiang, Z.-j. Jiang, X. Tian and W. Chen, J. Mater. Chem. A, 2014, 2, 441–450 CAS.
- S. Wang, D. Yu, L. Dai, D. W. Chang and J.-B. Baek, ACS Nano, 2011, 5, 6202–6209 CrossRef CAS PubMed.
- S. Wang, D. Yu and L. Dai, J. Am. Chem. Soc., 2011, 133, 5182–5185 CrossRef CAS PubMed.
- M. Chen, D. Kumar, C.-W. Yi and D. W. Goodman, Science, 2005, 310, 291–293 CrossRef CAS PubMed.
- G. J. Hutchings, Chem. Commun., 2008, 1148–1164 RSC.
- J. W. Hong, M. Kim, Y. Kim and S. W. Han, Chem.–Eur. J., 2012, 18, 16626–16630 CrossRef CAS PubMed.
- S. Guo, S. Zhang, L. Wu and S. Sun, Angew. Chem., 2012, 124, 11940–11943 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra10746b |
| This journal is © The Royal Society of Chemistry 2014 |