
Acid promoted cyclodehydration of amino alcohols with amide acetal†
Soonho Hwang,
Heemin Park,
Yongseok Kwon and
Sanghee Kim*
College of Pharmacy, Seoul National University, Seoul 151-742, Korea. E-mail: pennkim@snu.ac.kr; Fax: +82-2-888-0649; Tel: +82-2-880-2487
First published on 6th November 2014
Abstract
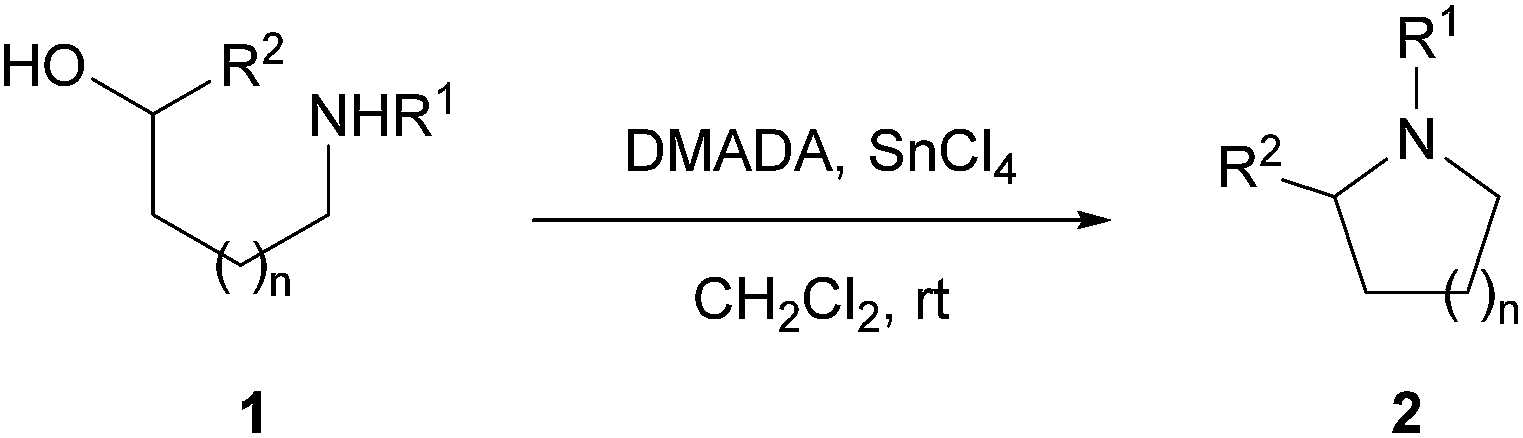
A convenient acid-promoted cyclization protocol for the formation of azaheterocycles from amino alcohols is described. The reaction involves the use of N,N-dimethylacetamide dimethyl acetal (DMADA) as the activating reagent of the hydroxyl group. Using this protocol, pyrrolidines or piperidines with various substituents can be synthesized in good to high yields.
Introduction
The formation of saturated azaheterocycles, such as pyrrolidine and piperidine, is of fundamental importance in organic synthesis, not only for the obvious reason that they are ubiquitous in nature,1 but also because they are key functional motifs in numerous synthetic molecules.2 As a result, there is an ever-growing number of synthetic methods, and the development of effective and environmentally benign methods is of considerable interest.3The intramolecular displacement of a hydroxyl group by an amine nucleophile is the most popular strategy to construct azaheterocycles.3 In this type of cyclization, the hydroxyl group requires prior activation for a smooth and high-yielding process. Conventionally, the hydroxyl group has been activated by converting it to a good leaving group such as a halide or sulfonate ester, which subsequently undergoes intramolecular N-alkylation under basic conditions. To reduce the number of chemical steps, direct methods were also devised, such as phosphorus assisted Mitsunobu-type reactions and Appel halogenation/in situ base-induced ring closure.4–6 However, there are some drawbacks such as the use of toxic reagents and the difficulty of byproduct removal. The direct ring closure of amino alcohols can also be achieved under acidic conditions.7 However, vigorous conditions such as strong acid and high temperature are needed to realize this type of transformation.
In our studies on the synthesis of azaheterocycle natural products and their analogues, we were confronted with the necessity to induce the cyclization of amino alcohols in neutral or mildly acidic conditions. To this end, we focused our attention to an oxocarbenium ion for the activation of the hydroxyl group. Although the chemistry of such carbenium ion has been well explored,8 its use as an activator of a hydroxyl group has not received much attention in substitution reactions,9,10 especially when an amine is used as the nucleophile.
As shown in Scheme 1, our cyclization strategy was based on the formation of an acetal intermediate A from amino alcohol 1 via trans-acetalization and the in situ generation of carbenium ion B for the activation of the hydroxyl group. Subsequent intramolecular displacement of the activated hydroxyl group by the amine nucleophile was envisioned to afford the azaheterocycle 2. For the successful implementation of our strategy, there are several issues to be addressed. The first is the complication associated with the trans-acetalization of the reagent with the hydroxyl group in the presence of the amino function. Despite the improbability of the chemoselective trans-acetalization, we were optimistic about the success of our cyclization strategy because the trans-acetalization is a process of thermodynamic equilibrium. Even in the case that only small proportion of A is formed compared to the N-acetalized intermediate C, the equilibrium would shift to make more A if that small amount undergoes an irreversible intramolecular cyclization readily. Another important issue is the ambident electrophilic reactivity of carbenium ion B. Although the preferred reactive site of such carbenium species is dependent on the reaction conditions and the nature of the nucleophile,8d we anticipated on the basis of entropic considerations that the carbenium function in B would react with the pendant amino group preferentially at the position of the sp3 carbon (path a) over the sp2 oxocarbenium carbon (path b) to yield azaheterocycles 2. Even in the case where D is formed as an initial reaction product due to the higher electrophilic reactivity of the sp2 oxocarbenium carbon, the azaheterocycle products 2 could be eventually formed through the reversible thermodynamic equilibrium processes.
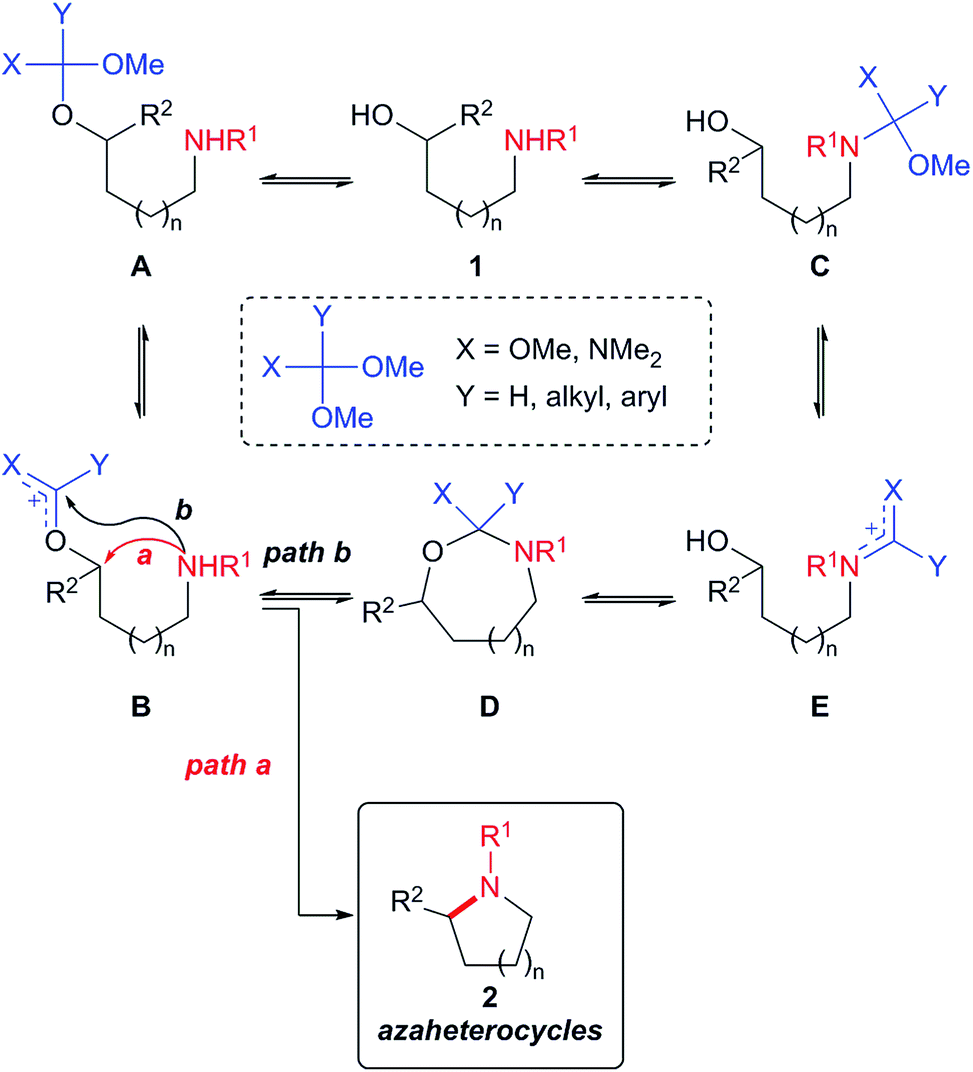 | ||
| Scheme 1 Strategy for the synthesis of azaheterocycles. | ||
Results and discussion
The simple amino alcohol 1a (Table 1) was chosen as the model substrate to test the viability of the envisioned cyclization process. At first, several types of carbenium ion precursors (2 equiv.) were screened in the presence of catalytic amount of HCl (0.1 equiv.)11 at the reflux temperature of 1,2-dichloroethane (1,2-DCE). When orthoesters were employed, such as trimethyl orthoacetate (TMOA) and trimethyl orthobenzoate (TMOB), the desired cyclized product was obtained, but only in low yield (entries 1 and 2). Substantial amounts of the N-acyl derivative and N,O-diacyl derivative of 1a were also formed.12 Alterations in the equivalent of reagents and acid, solvent, reaction time, and temperature failed to suppress the production of the N-acylated by-products.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Reagentb | Acid | Solvent | Temp (°C) | Time (h) | Yieldc (%) |
| a Reaction conditions: 1a (0.50 mmol), reagent (1.0 mmol), acid (0.050 mmol), solvent (10 mL) under N2.b Reagent for carbenium ion precursor.c Isolated yield of the HCl salt form of amine 2a.d 1 equiv. of HCl was used.e The values in parentheses indicate the yield of recovered starting material. CSA: (±)-camphorsulfonic acid. | ||||||
| 1 | TMOA | HCl | 1,2-DCE | Reflux | 18 | 35 |
| 2 | TMOB | HCl | 1,2-DCE | Reflux | 18 | 38 |
| 3 | DMFDA | HCl | 1,2-DCE | Reflux | 18 | 65 |
| 4 | DMADA | HCl | 1,2-DCE | Reflux | 18 | 85 |
| 5 | DMADA | HCld | CH2Cl2 | rt | 1 | 93 |
| 6 | DMADA | SnCl4 | CH2Cl2 | rt | 1 | 91 |
| 7 | DMADA | BF3·Et2O | CH2Cl2 | rt | 18 | 65 (12)e |
| 8 | DMADA | TiCl4 | CH2Cl2 | rt | 18 | 75 (8)e |
| 9 | DMADA | InBr3 | CH2Cl2 | rt | 18 | 75 (12)e |
| 10 | DMADA | CSA | CH2Cl2 | rt | 18 | 60 (23)e |
However, when amide acetals13 were employed, the reaction afforded the desired product without noticeable formation of the N-acylated compounds. N,N-Dimethylacetamide dimethyl acetal (DMADA)14 was much more efficient than N,N-dimethyl formamide dimethyl acetal (DMFDA) in promoting the reaction (entries 3 vs. 4). The use of DMADA led to 2a in 18 h in 85% yield under the screening conditions (0.1 equiv. of HCl, 1,2-DCE, reflux). The reaction with DMADA could proceed at room temperature with very satisfactory results, but it required more acid to go to completion (entry 5). A range of acids were screened at room temperature to further optimize this reaction and the typical results are shown in Table 1 (entries 6–10). SnCl4 was identified as the optimal choice for both chemical yield and reaction time. A catalytic amount of SnCl4 (0.1 equiv.) provided the product 2a after 1 h at room temperature in 91% yield. Many other Lewis acids produced the desired 2a, but did not fully complete the reaction at room temperature in spite of a long reaction time.
To verify the mechanism that we have postulated, an NMR study on the reaction of 1a with DMADA in CD2Cl2 was carried out.15 In the presence of catalytic amounts of SnCl4 (ref. 16) at room temperature, the 1H NMR spectrum revealed no predominant intermediate (Fig. S1, ESI†). The cyclized product 2a and N,N-dimethylacetamide (DMA) were generated in a nearly stoichiometric ratio.17 When the reaction mixture was quenched with water during the course of the reaction, we could detect the formation of the O-acetyl and N-acetyl derivatives, 3 and 4, in the same mutual ratio (Fig. S2, ESI†). These acetylated derivatives might arise by the action of water on the proposed carbenium intermediates (Scheme 2). This result together with the detection of DMA as a byproduct strongly suggested that the reaction proceeded via the envisaged intermediate carbenium ion species, such as I, and intramolecular nucleophilic attack of the amine.
 | ||
| Scheme 2 Plausible reaction mechanism. | ||
A carbenium ion intermediate similar to I has been proposed in the activation of the hydroxyl group with Vilsmeier reagent.10 Although several functional groups, such as thiols and imides, can serve as the nucleophile in the Vilsmeier reagent-mediated displacement of hydroxyl groups, an amine group has not been employed. We attempted the cyclization of 1a with Vilsmeier reagent to verify the benefit of DMADA (Table 2). When the reaction was performed at room temperature in the absence or presence of acid, the N-formyl chlorinated compound 5 was the major product of the reaction (entries 1–3). In the presence of base,18 the N-formyl chlorinated compound 5 was not formed. The cyclized compound 2a was the major component, but the yield was only 43% (entry 4). The reaction in THF afforded similar result (entry 5). At the reflux temperature of 1,2-DCE for 20 h, the yield of 2a increased up to 75% (entry 6), but ca. 20% of by-products were still formed such as O-formylated and N,O-diformylated derivatives of 1a. These results showed that the cyclization of amino alcohol with Vilsmieier reagent was much less efficient than that with DMADA with respect to reaction conditions and yield.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive (eq) | Solvent | Temp (°C) | Time (h) | Yieldb (%) | |
| 2a | 5 | |||||
| a Reaction conditions: 1a (0.50 mmol), Vilsmeier reagent (1.0 mmol), additive, solvent (10 mL) under N2.b Isolated yield of the HCl salt form of amine 2a and the amide 5. | ||||||
| 1 | — | CH2Cl2 | rt | 20 | 20 | 42 |
| 2 | HCl (1.0) | CH2Cl2 | rt | 20 | 25 | 43 |
| 3 | SnCl4 (0.1) | CH2Cl2 | rt | 20 | 22 | 47 |
| 4 | Et3N (3.0) | CH2Cl2 | rt | 20 | 43 | 0 |
| 5 | Et3N (3.0) | THF | rt | 20 | 42 | 0 |
| 6 | Et3N (3.0) | 1,2-DCE | Reflux | 20 | 75 | 0 |
With the optimized reaction conditions (DMADA and SnCl4), the cyclization of various types of substrates was investigated (Table 3). The cyclization reaction proceeded efficiently with various secondary amine substrates 1b–g to provide the N-substituted pyrrolidines 2b–g (entries 1–6). The reaction tolerated a broad range of N-substituted groups including alkyl, allyl, benzyl, and even sterically bulky groups. On the other hand, the reaction with the primary amine substrate 1h failed to yield the corresponding product and a complex mixture of unidentified products was obtained (entry 7). This failure is most likely because of the facile condensation of the primary amine with DMADA to give the acetamidine or imidate ester.19
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 1 | R1 | R2 | n | Time (h) | 2 | Yieldb (%) |
| a Reaction conditions: 1 (0.50 mmol), DMADA (1.0 mmol), SnCl4 (0.050 mmol), CH2Cl2 (10 mL), rt.b Isolated yield of the HCl salt form of 2.c Cyclized product was not detected.d The reaction was conducted in 1,2-DCE (5 mL) at reflux temperature with 100 wt% of 4 Å molecular sieve. | |||||||
| 1 | 1b | 4-OMe–Bn | H | 1 | 1 | 2b | 92 |
| 2 | 1c | 4-CF3–Bn | H | 1 | 1 | 2c | 90 |
| 3 | 1d | Allyl | H | 1 | 1 | 2d | 90 |
| 4 | 1e | C7H15 | H | 1 | 1 | 2e | 92 |
| 5 | 1f | 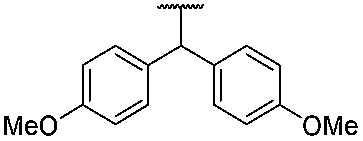 |
H | 1 | 1 | 2f | 91 |
| 6 | 1g | 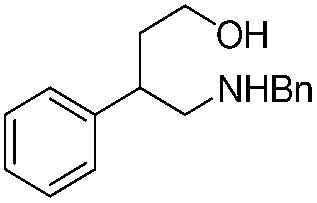 |
1 | 2g | 93 | ||
| 7 | 1h | H | H | 1 | 16 | 2h | —c |
| 8 | 1i | Bn | H | 2 | 2 | 2i | 90 |
| 9 | 1j | 4-OMe–Bn | H | 2 | 2 | 2j | 91 |
| 10 | 1k | C7H15 | H | 2 | 2 | 2k | 89 |
| 11 | 1l | Bn | H | 3 | 16 | 2l | —c |
| 12 | 1m | Bn | Me | 1 | 5 | 2m | 89 |
| 13d | 1n | Bn | n-Bu | 1 | 16 | 2n | 82 |
| 14d | 1o | Bn | Ph | 1 | 16 | 2o | 82 |
Under these reaction conditions, the six-membered piperidine ring formation could also be achieved with high yields (entries 8–10). However, the formation of a seven-membered azepane ring was not successful even when exposed to prolonged high temperature (entry 11). The substrates with a secondary hydroxyl group were also well suited for the reaction and gave the corresponding cyclized products (entries 12–14). However, this type of substrates generally required a longer reaction time and higher temperature to reach completion. We considered this reaction proceeds via SN2 mechanism because the chiral non-racemic 1o afforded the corresponding cyclized product without any racemization.15
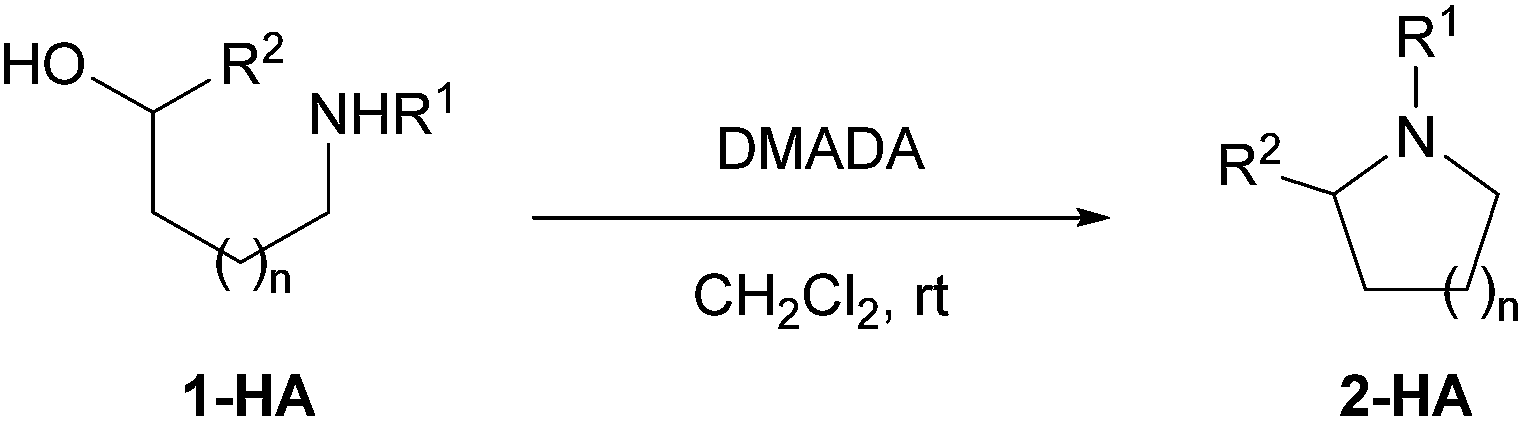
Because the cyclized product could be obtained even with a stoichiometric equivalent of HCl (Table 1, entry 5), we anticipated that the acid salt of the amino alcohol itself could undergo the reaction with DMADA without needing the extra acid catalyst. The success of such transformation would open avenues for simple manipulation of the amino alcohol salt substrates because it does not require the addition of base to liberate the free amine function. As we expected, when the HCl or TFA salt form of amino alcohol 1 was employed as a substrate and merely treated with DMADA, the cyclized product 2 was successfully obtained in high yields (Table 4).
|
||||
|---|---|---|---|---|
| Entry | 1-HA | Time (h) | 2-HA | Yieldb (%) |
| a Reaction conditions: 1-HA (0.50 mmol), DMADA (1.0 mmol), CH2Cl2 (10 mL), rt.b Isolated yield of 2-HA.c The reaction was conducted in 1,2-DCE (5 mL) at reflux temperature with 100 wt% of 4 Å molecular sieve. | ||||
| 1 | 1a-HCl | 1 | 2a-HCl | 93 |
| 2 | 1a-TFA | 1 | 2a-TFA | 94 |
| 3 | 1b-HCl | 1 | 2b-HCl | 91 |
| 4 | 1b-TFA | 1 | 2b-TFA | 90 |
| 5 | 1c-HCl | 1 | 2c-HCl | 94 |
| 6 | 1d-HCl | 1 | 2d-HCl | 93 |
| 7 | 1i-HCl | 16 | 2i-HCl | 94 |
| 8 | 1i-TFA | 2 | 2i-TFA | 91 |
| 9 | 1j-HCl | 2 | 2j-HCl | 91 |
| 10 | 1k-HCl | 2 | 2k-HCl | 90 |
| 11c | 1m-HCl | 16 | 2m-HCl | 89 |
| 12c | 1n-HCl | 5 | 2n-HCl | 85 |
Building on the above results, an efficient process for converting N-Boc protected amino alcohols to the corresponding azaheterocycles was developed. For this, we employed the known N-Boc protected amino alcohol 6 (Scheme 3) which has been converted to the natural product crispine A (7) in several ways.20
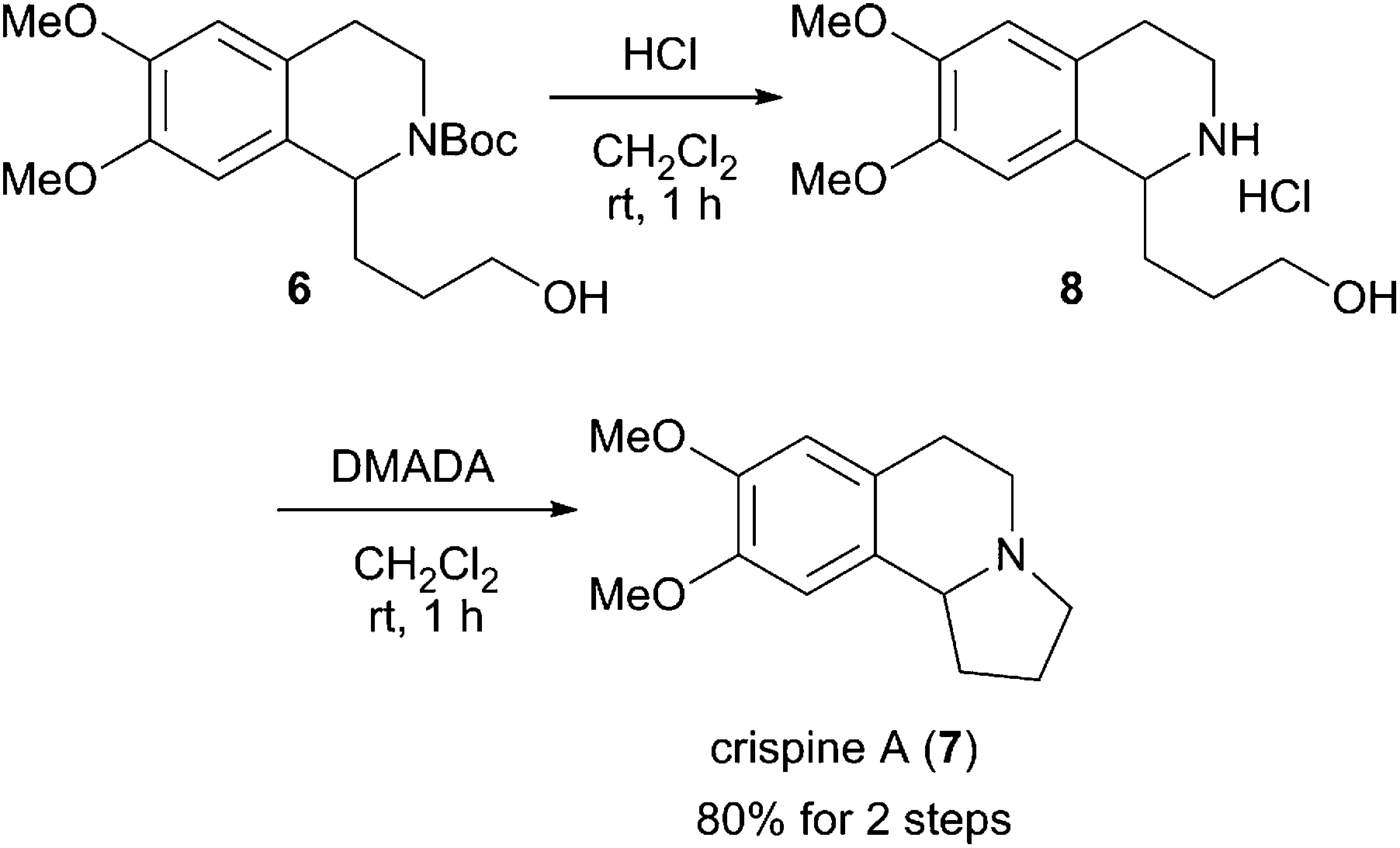 | ||
| Scheme 3 Synthesis of crispine A (7). | ||
As the first step, the Boc group was removed by using HCl in CH2Cl2, and the hydrochloride salt 8 was obtained by evaporation. Without further purification, salt 8 was next treated with DMADA at room temperature for 1 h. This reaction sequence gave a high yield of the cyclization product, crispine A (7), which gave spectral data that was identical to what previously reported.20,21
Conclusions
In conclusion, we have developed a convenient and efficient synthetic method for the generation of azaheterocycles from amino alcohols. Our method is unique among other previously developed synthetic methods in that the cyclization is promoted by acid under mild conditions. This method is applicable to substrates with various functional groups, especially those with base-sensitive groups. A key feature of this protocol is the use of an aza-oxo-stabilized carbenium ion for the activation of the hydroxyl group which has rarely been explored in substitution reactions, especially when an amine used as the nucleophile. Several types of pyrrolidines and piperidines were successfully prepared in good to high yields from the amino alcohols with DMADA in the presence of an acid catalyst. Moreover, we found that the acid salt of the amino alcohol could undergo the facile reaction with DMADA without requiring any additional acid catalyst. These results open avenues for the mild synthesis of azaheterocycles with the advantages of simple manipulations, especially when the amino alcohol substrate is in its salt form.Experimental
General information
All chemicals were of reagent grade and used as received. All reactions were performed under dry nitrogen using distilled, dry solvents. The reactions were monitored by TLC (Merck®, Silica gel 60 F254). Flash column chromatography was performed on silica gel (230–400 mesh). 1H (300 or 400 MHz) and 13C NMR (75 or 100 MHz) spectra were recorded. Chemical shifts (δ) are reported in ppm relative to the non-deuteriated solvent as internal reference; coupling constants (J) are given in Hz. Multiplicities are denoted as follows: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. The 1H NMR spectra are presented as follows: chemical shift (multiplicity, coupling constant, integration). IR spectra were recorded with a Fourier transform infrared spectrometer. High resolution mass spectra (HRMS) were obtained by electron ionization (EI) using a double focusing mass spectrometer. Previously reported compounds were confirmed by comparison of their 1H NMR with those of references.Representative procedure for the synthesis of amino alcohol 1a (reductive amination protocol)
Benzaldehyde (6.9 mL, 67.3 mmol, 1.2 equiv.) and sodium sulfate (Na2SO4, 15 g, 1.9 equiv.) were added to a solution of commercially available 4-amino-1-butanol (5.0 g, 56.1 mmol, 1 equiv.) in CH2Cl2 (56 mL, 1.0 M) under N2 atmosphere. After stirring for 15 h at room temperature, the crude reaction mixture was filtered through a pad of Celite, and the filtrate was concentrated under reduced pressure. The crude mixture obtained was dissolved in EtOH (112 mL, 0.5 M) and NaBH4 (4.3 g, 112.2 mmol, 2 equiv.) was added portion wise at 0 °C. After stirring for an additional 2 h at room temperature, the reaction was quenched by the addition of saturated NH4Cl solution at 0 °C. After evaporation of the excess EtOH, the reaction mixture was diluted with water and extracted twice with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (CH2Cl2–MeOH, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 + 1% NH4OH) to give known N-Bn amino alcohol 1a.22
:1 + 1% NH4OH) to give known N-Bn amino alcohol 1a.22
Amino alcohols 1b,23 1c, 1e, 1i,22 1j, 1k,24 and 1l22 were prepared by this reductive amination protocol with 11.2 mmol of corresponding amines. The following requisite aldehydes (p-anisaldehyde, 4-(trifluoromethyl)benzaldehyde, and heptaldehyde) and amines (4-amino-1-butanol, 5-amino-1-pentanol and 6-amino-1-hexanol) were commercially available.
Representative procedure for the synthesis of amino alcohol 1d (amidation – LAH reduction protocol)
Allylamine (2.3 mL, 30 mmol, 1.5 equiv.) and γ-butyrolactone (1.6 mL, 20 mmol, 1 equiv.) were dissolved in benzene (10 mL, 2.0 M) and refluxed for 12 h under a N2 atmosphere. After cooling to room temperature, excess allylamine and benzene were removed under reduced pressure. The residue was diluted with EtOAc, washed with a 1.0 M HCl solution and brine, dried over MgSO4, and concentrated in vacuo to give N-allyl-4-hydroxybutanamide, which was used in the next step without further purification. To a solution of LiAlH4 (1.5 g, 40 mmol, 2 equiv.) in THF (80 mL) at 0 °C was added slowly dropwise a THF (20 mL) solution of the amide (2.86 g, 20 mmol, 1 equiv.). After stirring for 30 min at room temperature, the reaction mixture was refluxed for 18 h. After cooling to 0 °C, the reaction was quenched by the careful addition of H2O (1.5 mL), 10% NaOH solution (1.5 mL), and H2O (4.5 mL) sequentially. After stirring an additional 2 h at room temperature, the crude mixture was filtered through a pad of Celite, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (CH2Cl2–MeOH, 10:1 + 1% NH4OH) to give known N-allyl amino alcohol 1d.22
Amino alcohols 1f, 1g, 1m,25 1n, and 1o26 were prepared by this amidation – LAH reduction protocol with 4 mmol of corresponding lactones. The following requisite lactones (γ-valerolactone, γ-octanolactone, and γ-phenyl-γ-butyrolactone) and amines (4,4′-dimethoxybenzhydrylamine and benzylamine) were commercially available. α-Phenyl-γ-butyrolactone required for 1g was prepared by a previously developed procedure.27
Representative procedure for the preparation of amino alcohol salts 1-HCl
HCl solution (4.0 M soln in dioxane, 5 equiv.) was slowly added to a solution of the obtained amino alcohol 1 (1 equiv.) in THF (0.3 M) at 0 °C. After stirring for an additional 5 min, the crude reaction mixture was concentrated under reduced pressure. The crude solid was purified by recrystallization (EtOAc and hexane) to afford pure 1-HCl.:2, 0.80 mL, molar ratio) and Noyori's transfer hydrogenation catalyst RuCl(p-cymene)[(S,S)-Ts-DPEN] (10 mg, 1 mol %) at room temperature. The resulting solution was stirred at room temperature for 24 h. The reaction mixture was diluted with water and extracted twice with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexane–EtOAc, 1:1) to give N-Bn amide alcohol as white solid (330 mg, 82%). 1H NMR (300 MHz, CDCl3) δ 1.97–2.16 (m, 2H), 2.37 (t, J = 7.1 Hz, 2H), 3.48 (brs, 1H), 4.42 (d, J = 5.5 Hz, 2H), 4.77 (dd, J = 7.5 Hz, 4.6 Hz, 1H), 6.12 (brs, 1H), 7.21–7.35 (m, 10H). The 1H NMR spectrum was identical with that reported for its racemate.29To a solution of LiAlH4 (125 mg, 3.3 mmol, 3 equiv.) in THF (20 mL) at 0 °C was added slowly dropwise a THF (5 mL) solution of the N-Bn amide alcohol (296 mg, 1.1 mmol, 1 equiv.). After stirring for 30 min at room temperature, the reaction mixture was refluxed for 18 h. After cooling to 0 °C, the reaction was quenched by the careful addition of H2O (125 μL), 10% NaOH solution (125 μL), and H2O (375 μL) sequentially. After stirring an additional 2 h at room temperature, the crude mixture was filtered through a pad of Celite, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (CH2Cl2–MeOH, 10:1 + 1% NH4OH) to give non-racemic amino alcohol 1o as white solid (265 mg, 85%). 1H NMR (300 MHz, CDCl3) δ 1.56–1.74 (m, 2H), 1.76–1.87 (m, 1H), 1.89–1.99 (m, 1H), 2.66 (qd, J = 4.1 Hz, 12.0 Hz, 1H), 2.81 (qd, J = 3.8 Hz, 11.6 Hz, 1H), 3.80 (s, 2H), 4.68 (dd, J = 3.1 Hz, 8.2 Hz, 1H), 7.14–7.37 (m, 10H). The 1H NMR spectrum was identical with that reported for its racemate.26 The optical purity (91% ee) of non-racemic amino alcohol 1o was determined by 1H NMR spectroscopic analysis of N-Boc (S)-(+)-α-(trifluoromethyl)phenylacetyl ester derivative.
Representative procedure for the SnCl4-catalyzed cyclization of amino alcohols 1 (Table 2)
To a stirred solution of amino alcohol 1 (0.50 mmol, 1 equiv.) in CH2Cl2 (0.05 M, 10 mL) was added SnCl4 (1.0 M soln in CH2Cl2, 50 μL, 0.050 mmol, 0.1 equiv.) and N,N-dimethylacetamide dimethyl acetal (DMADA, 0.17 mL, 1.0 mmol, 2 equiv.) at room temperature. After disappearance of the starting material, HCl (4.0 M soln in dioxane, 0.50 mL, 2.0 mmol, 4 equiv.) was added to make 2 into its corresponding salt. After stirring an additional 5 min at room temperature, the crude mixture was azeotropically evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (CH2Cl2–MeOH, 10:1) to give 2-HCl.30 After checking the chemical yield, small amounts of 2-HCl was basified with a NaOH solution to confirm the chemical identity.
Representative procedure for the cyclization of amino alcohol salts 1-HA (Table 3)
To a stirred solution of 1-HA (0.50 mmol, 1 equiv.) in CH2Cl2 (0.05 M, 10 mL) was added DMADA (0.17 mL, 1.0 mmol, 2 equiv.) at room temperature. The following reaction procedure was the same as the above procedure.Procedure for capturing O-acetyl derivative 3 and N-acetyl derivative 4
To a stirred solution of 1a (0.50 mmol, 1 equiv.) in CH2Cl2 (0.05 M, 10 mL) was added SnCl4 (0.1 M soln in CH2Cl2, 50 μL, 0.0050 mmol, 0.01 equiv.) and DMADA (0.17 mL, 1.0 mmol, 2 equiv.) at room temperature and continued to stir for 30 min. After the mixture was cooled to 0 °C, the reaction was quenched with water and extracted with EtOAc. The organic layer was washed with saturated NaHCO3 solution and brine, dried with MgSO4, and concentrated in vacuo. The resulting residue was purified by flash chromatography on silica gel (CH2Cl2–MeOH, 15:1 + 1% NH4OH) to give 3 and 4.
Representative procedure for the cyclization of amino alcohol 1a using Vilsmeier reagent (Table 2)
To a stirred solution of amino alcohol 1a (0.50 mmol, 1 equiv.) in solvent (0.05 M, 10 mL) was added additive and Vilsmeier reagent, easily prepared from dimethyl formamide and oxalyl chloride,10b (1.0 mmol, 2 equiv.) at room temperature. The reaction mixture was stirred at the indicated temperature for the indicated amount of time. After disappearance of the starting material, HCl (4.0 M soln in dioxane, 0.50 mL, 2.0 mmol, 4 equiv.) was added to make 2a into its corresponding salt. After stirring an additional 5 min at room temperature, the crude mixture was azeotropically evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (CH2Cl2–MeOH, 10:1) to give 2a-HCl and 5.
:1 + 1% NH4OH) to give crispine A (7) (93.2 mg, 80% for 2-steps) as a colorless solid. 1H NMR (300 MHz, CDCl3) δ 1.63–1.76 (m, 1H), 1.81–1.99 (m, 2H), 2.25–2.35 (m, 1H), 2.52–2.74 (m, 3H), 2.92–3.09 (m, 2H), 3.12–3.19 (m, 1H), 3.43 (t, J = 8.2 Hz, 1H), 3.82 (s, 6H), 6.54 (s, 1H), 6.58 (s, 1H).Acknowledgements
This work was supported by the Mid-Career Researcher Program (no. 2013R1A2A1A01015998) of the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP).Notes and references
- (a) Modern Alkaloids. Structure, Isolation, Synthesis and Biology, ed. E. Fattorusso and O. Taglialatela-Scafati, Wiley-VCH Verlag, Weinheim, Germany, 2008 Search PubMed; (b) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435 RSC; (c) A. R. Pinder, Nat. Prod. Rep., 1986, 3, 171 RSC.
- C. W. Bird, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon, Oxford, 1996, vol. 2, ch. 19, pp. 933–967 Search PubMed.
- For reviews, see: (a) N. R. Candeias, L. C. Branco, P. M. P. Gois, C. A. M. Afonso and A. F. Trindade, Chem. Rev., 2009, 109, 2703 CrossRef CAS PubMed; (b) A. Mitchinson and A. Nadin, J. Chem. Soc., Perkin Trans. 1, 2000, 17, 2862 RSC; (c) M. G. P. Buffat, Tetrahedron, 2004, 60, 1701 CrossRef CAS PubMed; (d) P. M. Weintraub, J. S. Sabol, J. M. Kane and D. R. Borcherding, Tetrahedron, 2003, 59, 2953 CrossRef CAS; (e) M. Pichon and B. Figadère, Tetrahedron: Asymmetry, 1996, 7, 927 CrossRef CAS; (f) R. C. Larock, in Comprehensive Organic Transformations, Wiley-VCH, New York, 2nd edn, 1999, pp. 689–702 and 779–784 Search PubMed.
- For a leading reference and some recent examples of Mitsunobu-type cyclizations, see: (a) R. C. Bernotas and R. V. Cube, Tetrahedron Lett., 1991, 32, 161 CrossRef CAS; (b) H. Wong, E. C. Garnier-Amblard and L. S. Liebeskind, J. Am. Chem. Soc., 2011, 133, 7517 CrossRef CAS PubMed; (c) N. Saha, T. Biswas and S. K. Chattopadhyay, Org. Lett., 2011, 13, 5128 CrossRef CAS PubMed; (d) Y. Ukaji, M. Imai, T. Yamada and K. Inomata, Heterocycles, 2000, 52, 563 CrossRef CAS.
- For some recent examples of Appel-type cyclizations, see: (a) G. Archibald, C.-P. Lin, P. Boyd, D. Barker and V. Caprio, J. Org. Chem., 2012, 77, 7968 CrossRef CAS PubMed; (b) J.-J. Zhuang, J.-L. Ye, H.-K. Zhang and P.-Q. Huang, Tetrahedron, 2012, 68, 1750 CrossRef CAS PubMed; (c) H. Yoon, K. S. Cho and T. Sim, Tetrahedron: Asymmetry, 2014, 25, 497 CrossRef CAS PubMed.
- For other approaches for direct cyclization of amino alcohols, see: (a) D. Pingen and D. Vogt, Catal. Sci. Technol., 2014, 4, 47 RSC; (b) P. H. Huy and A. M. P. Koskinen, Org. Lett., 2013, 15, 5178 CrossRef CAS PubMed; (c) F. Xu, B. Simmons, R. A. Reamer, E. Corley, J. Murry and D. Tschaen, J. Org. Chem., 2008, 73, 312 CrossRef CAS PubMed; (d) R. M. de Figueiredo, R. Fröhlich and M. Christmann, J. Org. Chem., 2006, 71, 4147 CrossRef CAS PubMed.
- (a) H. Tanaka, Y. Murakami, T. Aizawa and S. Torii, Bull. Chem. Soc. Jpn., 1989, 62, 3742 CrossRef CAS; (b) C. Papageorgiou and X. Borer, Helv. Chim. Acta, 1988, 71, 1079 CrossRef CAS.
- For reviews, see: (a) R. R. Naredla and D. A. Klumpp, Chem. Rev., 2013, 113, 6905 CrossRef CAS PubMed; (b) P. J. Stang, in Carbocation Chemistry, ed. G. A. Olah and G. K. S. Prakash, Wiley: Hoboken, NJ, 2004, pp. 1–6 Search PubMed; (c) H. Grützmacher and C. M. Marchand, Coord. Chem. Rev., 1997, 163, 287 CrossRef; (d) U. Pindur, J. Müller, C. Flo and H. Witzel, Chem. Soc. Rev., 1987, 16, 75 RSC.
- (a) H. Neumann, Chimia, 1969, 23, 267 CAS; (b) R. G. Harvey, S. H. Goh and C. Cortez, J. Am. Chem. Soc., 1975, 97, 3468 CrossRef CAS; (c) H. C. Kolb and K. B. Sharpless, Tetrahedron, 1992, 48, 10515 CrossRef CAS; (d) T. Zheng, R. S. Narayan, J. M. Schomaker and B. J. Borhan, J. Am. Chem. Soc., 2005, 127, 6946 CrossRef CAS PubMed.
- (a) P. A. Procopiou, A. C. Brodie, M. J. Deal and D. F. Hayman, Tetrahedron Lett., 1993, 34, 7483 CrossRef CAS; (b) A. G. M. Barrett, D. C. Braddock, R. A. James, N. Koike and P. A. Procopiou, J. Org. Chem., 1998, 63, 6273 CrossRef CAS PubMed; (c) Y. Kawano, N. Kaneko and T. Mukaiyama, Chem. Lett., 2005, 34, 1612 CrossRef CAS.
- Commercially available HCl solution (4.0 M in dioxane) was used.
- R. A. Swaringen Jr, J. F. Eaddy and T. R. Henderson, J. Org. Chem., 1980, 45, 3986 CrossRef.
- For review, see: R. F. Abdulla and R. S. Brinkmeyer, Tetrahedron, 1979, 35, 1675 CrossRef CAS.
- Commercial DMADA generally contains 5 to 10 percent methanol as a stabilizer.
- For more details, see the Experimental section and ESI.†.
- Only 0.01 equiv. of SnCl4 was used to slow down the reaction.
- Minor amounts of methyl acetate and dimethylamine were also detected.
- Vilsmeier reagent-mediated substitution reaction is usually performed under basic conditions. For examples, see ref. 10.
- J. R. Harjani, C. Liang and P. G. Jessop, J. Org. Chem., 2011, 76, 1683 CrossRef CAS PubMed.
- (a) M. Miyazaki, N. Ando, K. Sugai, Y. Seito, H. Fukuoka, T. Kanemitsu, K. Nagata, Y. Odanaka, K. T. Nakamura and T. Itoh, J. Org. Chem., 2011, 76, 534 CrossRef CAS PubMed; (b) E. Forró, L. Schönstein and F. Fülöp, Tetrahedron: Asymmetry, 2011, 22, 1255 CrossRef PubMed; (c) N. S. S. Reddy, B. J. M. Reddy and B. V. S. Reddy, Tetrahedron Lett., 2013, 54, 4228 CrossRef CAS PubMed.
- Q. Zhang, G. Tu, Y. Zhao and T. Cheng, Tetrahedron, 2002, 58, 6795 CrossRef CAS.
- Y.-H. Wang, J.-L. Ye, A.-E. Wang and P.-Q. Huang, Org. Biomol. Chem., 2012, 10, 6504 CAS.
- W. F. McCalmont, J. R. Patterson, M. A. Lindenmuth, T. N. Heady, D. M. Haverstick, L. S. Gray and T. L. Macdonald, Bioorg. Med. Chem., 2005, 13, 3821 CrossRef CAS PubMed.
- A. Gelain, I. Bettinelli, D. Barlocco, B.-M. Kwon, T.-S. Jeong, K.-H. Cho and L. Toma, J. Med. Chem., 2005, 48, 7708 CrossRef CAS PubMed.
- M. Scheideman, G. Wang and E. Vedejs, J. Am. Chem. Soc., 2008, 130, 8669 CrossRef CAS PubMed.
- H.-C. Zhang, B. D. Harris, M. J. Costanzo, E. C. Lawson, C. A. Maryanoff and B. E. Maryanoff, J. Org. Chem., 1998, 63, 7964 CrossRef CAS.
- C. Liu, C. He, W. Shi, M. Chen and A. Lu, Org. Lett., 2007, 9, 5601 CrossRef CAS PubMed.
- S. P. Webster, M. Binnie, K. M. M. McConnell, K. Sooy, P. Ward, M. F. Greaney, A. Vinter, T. D. Pallin, H. J. Dyke, M. I. A. Gill, I. Warner, J. R. Seckl and B. R. Walker, Bioorg. Med. Chem. Lett., 2010, 20, 3265 CrossRef CAS PubMed.
- G. M. Coppola and R. E. Damon, J. Heterocycl. Chem., 1995, 32, 1133 CrossRef CAS.
- Due to somewhat volatile property of amine 2, product was obtained as its HCl salt form.
- W. Zhang, X. Dong and W. Zhao, Org. Lett., 2011, 13, 5386 CrossRef CAS PubMed.
- G. A. Molander, P. E. Gormisky and D. L. Sandrock, J. Org. Chem., 2008, 73, 2052 CrossRef CAS PubMed.
- X. Zhao, D. Liu, H. Guo, Y. Liu and W. Zhang, J. Am. Chem. Soc., 2011, 133, 19354 CrossRef CAS PubMed.
- M. C. Venuti and O. Ort, Synthesis, 1988, 12, 985 CrossRef.
- O. Tomashenko, V. Sokolov, A. Tomashevskiy, H. A. Buchholz, U. Welz-Biermann, V. Chaplinski and A. de Meijere, Eur. J. Org. Chem., 2008, 30, 5107 CrossRef.
- K. Fujita, T. Fujii and R. Yamaguchi, Org. Lett., 2004, 6, 3525 CrossRef CAS PubMed.
- L. Wu, I. Fleischer, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2013, 135, 3989 CrossRef CAS PubMed.
- L. D. Julian and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 13813 CrossRef CAS PubMed.
- K.-J. Xiao, Y. Wang, K.-Y. Ye and P.-Q. Huang, Chem.–Eur. J., 2010, 16, 12792 CrossRef CAS PubMed.
- G. Belanger, R. Larouche-Gauthier, F. Menard, M. Nantel and F. Barabe, J. Org. Chem., 2006, 71, 704 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra of all new compounds, 1H NMR spectroscopy study, and stereochemical proofs. See DOI: 10.1039/c4ra10625c |
| This journal is © The Royal Society of Chemistry 2014 |