
High redox activity of Sr-substituted lanthanum manganite perovskites for two-step thermochemical dissociation of CO2†
Antoine Demont and
Stéphane Abanades*
Processes, Materials, and Solar Energy Laboratory, PROMES-CNRS (UPR 8521), 7 Rue du Four Solaire, 66120 Font-Romeu, France. E-mail: stephane.abanades@promes.cnrs.fr; Tel: +33 (0)468307730
First published on 20th October 2014
Abstract
The La1−xSrxMnO3−δ series of non-stoichiometric perovskites (x = 0.35, 0.50, 0.65, 0.80) was examined in the context of solar-driven two-step thermochemical dissociation of CO2. Powder X-ray diffraction and thermochemical performance characterization were performed in order to assess the redox activity of these materials toward thermal reduction under inert atmosphere followed by re-oxidation for CO generation from CO2. To a certain extent, controlled introduction of Sr2+ into LaMnO3 allowed tuning the redox thermodynamics within the series, thus resulting in high activity toward both thermal reduction and CO2 dissociation. La0.50Sr0.50MnO3−δ composition appeared to be the most suitable trade-off for thermochemical CO2 splitting. Maximum CO production of about 270 μmol g−1 was reached during the CO2 splitting step with an optimal re-oxidation temperature of 1050 °C (after thermal reduction under Ar at 1400 °C), although the re-oxidation yield was limited to around 50%. Decreasing the amount of substituted Sr enhanced the re-oxidation yield at the expense of a lower final reduction extent, thus lowering the global amount of produced CO. The evolution of the Mn oxidation state implied partial re-oxidation of Mn3+ into Mn4+, thereby confirming the activation of Mn4+/Mn3+ redox pair in the perovskites. An elevated electronic transfer occurred within the Mn4+/Mn3+ redox pair (superior to that involved in the case of ceria within the Ce4+/Ce3+ redox pair), showing that mixed valence perovskites have clear potential for displaying redox properties suitable for efficient solar-driven thermochemical CO2 dissociation.
Introduction
Within the context of solar fuels production, the recycling and valorization of carbon dioxide into carbon monoxide, one of the main component of syngas, has emerged as a promising solution, as CO can later be combined with H2 for the generation of synthetic fuels. Over the past decades, several multistep processes have been envisioned for an efficient realization of such concept.1–3 Among these, the solar thermochemical dissociation of CO2 is an attractive one, benefiting from both full utilization of the solar spectrum and ease of implementation.4 Since the direct thermolysis reaction (2CO2 → 2CO + O2) occurs at temperatures that are not practically achievable in a solar reactor designed for large scale production, such a concept greatly relies on the redox thermodynamics of metal or mixed-metal oxides, that catalyze the CO2 dissociation when applying two-step thermochemical treatments. Generally, the two-step process firstly consists of the thermal reduction of the oxide material (endothermic step) at temperature T1, associated with oxygen liberation from the bulk catalyst. Secondly, re-oxidation is performed at T2 lower than T1 (exothermic step) under carbon dioxide exposure, which promotes CO2 splitting and subsequent CO generation, owing to oxygen capture by the formerly reduced catalyst. Decreasing T1 while retaining efficient CO production is currently one of the main crucial challenges in the field of solar fuels, in order to ease the design and operation of large scale facilities involving solar tower receivers. Historically, stoichiometric oxides were primarily considered for solar fuel generation (involving CO or H2 production) by two-step thermochemical cycles,5–13 until the efficiency of non-stoichiometric compounds such as ceria was demonstrated.4,14–19 For non-stoichiometric oxides, two-step thermochemical dissociation of CO2 can be summarized with the following equations:Step 1 (reduction, O2 generation): AOx → AOx−δ + δ/2O2 (T1, endothermic step).
Step 2 (re-oxidation, CO generation): AOx−δ + δCO2 → AOx + δCO (T2 < T1, exothermic step).
Advantages encountered when dealing with non-stoichiometric oxides mainly lie on two characteristics. On the one hand, in contrast to stoichiometric compounds, their reduction and subsequent re-oxidation is based on continuous infinitesimal variations of the oxygen content and metal oxidation state. Such infinitesimal changes require less energy input than the total reduction of the metallic cations, enabling to trigger the reduction at lower temperatures. CeO2 provides an archetypal example of this behavior, as the partial reduction into the non-stoichiometric form CeO2−δ (where δ is the number of oxygen vacancies per formula unit) is engaged below 1100 °C without any phase change,20 whereas total reduction of CeO2 into the stoichiometric form Ce2O3 occurs at much higher temperature (2000 °C). On the other hand, non-stoichiometry is often associated with crystal structure flexibility. This allows multiple chemical substitutions to be performed into the considered compounds, offering large perspectives for the tuning of the properties. The flexibility also results in a rich defect chemistry: for example, CeO2−δ has the ability to cope with a large amount of oxygen vacancies while retaining the fluorite structure. This structural mechanism supplies pathways for the diffusion of oxygen atoms throughout the material, resulting in highly mobile O2− species that can be delocalized from an oxygen-filled site to an oxygen vacant site within the crystal network. This characteristic is beneficial for rapid homogenization of the bulk catalyst oxygen content, which is required during the solid–gas reactions.
Thus, based on the case of the fluorite-type CeO2 and various substituted versions of this compound, there has been increased recognition that non-stoichiometric compounds could be used to efficiently produce solar fuels via two-step thermochemical cycles. Despite the fact that perovskite compounds are a prototypical example of materials displaying a rich defect chemistry, easily enabling the formation of oxygen vacancies in their crystal structures, these materials have long received minor attention in the field when compared to fluorite-based materials. Within a process comparable to two-step thermochemical dissociation of CO2, several studies have demonstrated that CH4 to syngas conversion was catalyzed by the La1−xSrxMnO3−δ series.21–23 Scheffe et al. have theoretically validated the use of selected perovskites by gathering existing thermodynamic data on LaMnO3 based compounds. Their work was experimentally supported by the observation of redox activity associated with CO2 splitting using La1−xSrxMnO3−δ (x = 0.35).24 Mc Daniel et al. have established that significant H2 and CO thermochemical productions could be achieved with LaAlO3-based perovskites by performing partial reduction at 1350 °C,25 a temperature that is slightly lower than those commonly employed with ceria (1400–1500 °C). Even more recently, significant H2 productions were reported with perovskites that were thermally reduced at temperatures as low as 1000 °C,26 while Sr-substituted LaMnO3 based monoliths were shown to generate large H2 quantities during two-step thermochemical water splitting.27 Given the rich perspectives offered by perovskites in the context of solar CO2 splitting, we have undertaken the study of La1−xSrxMnO3−δ for a large range of Sr contents (x = 0.35 to x = 0.80). Powder X-ray diffraction (PXRD) was used to ensure phase purity and validate the crystal structures, after which the materials were characterized to determine their thermochemical redox performances. A detailed study of the redox behavior of the compounds during both the thermal reduction and the CO2 splitting was performed, giving insights on the conditions required for achieving significant CO productions with these materials.
Experimental
Polycrystalline samples of La0.65Sr0.35MnO3−δ (LSM35), La0.50Sr0.50MnO3−δ (LSM50), La0.35Sr0.65MnO3−δ (LSM65), and La0.20Sr0.80MnO3−δ (LSM80) were prepared by the conventional solid state method. Stoichiometric amounts (in term of cationic composition) of La2O3, SrCO3 and MnO2 were weighted and intimately mixed within an agate mortar and pestle. La2O3 was calcined at 950 °C prior to utilization in order to eliminate La(OH)3. The powders were introduced into an alumina crucible and fired at 1000 °C in air for 6 h. After regrinding, the poorly sintered samples were fired at 1450 °C (LSM35 and LSM50) or 1500 °C (LSM65 and LSM80) in air for 6 h. This final step of the synthesis was reproduced until phase purity was observed by powder X-ray diffraction (PXRD). Phase identification was carried out by PXRD analysis with data collected at room temperature using an X'Pert Pro PANalytical diffractometer equipped with an X'celerator detector and working with Cu Kα1 and Kα2 radiations (λ = 0.15418 nm). The X-ray diffraction measurements of θ–θ symmetrical scans were made in the range 10–100°. The step size and the time per step were respectively fixed at 0.01° and 5 s. Crystalline phases were identified by comparison with standard reference patterns (Powder diffraction file PDF-2, International Centre for Diffraction Data, ICDD). Unit cell parameters determination was carried out with LeBail fits that were performed with the Fullprof software28 included in the Winplotr package.29 Thermogravimetric (TG) measurements were carried out using a SETARAM Sestys Evolution device, with approximately 120 mg of sample powder placed in a platinum crucible. The thermal reduction step was carried out under an Ar flow of 20 ml min−1 (2 ppm O2), and CO2 was injected for the CO2 dissociation step (CO2 mole fraction of 50% in Ar). All heating and cooling steps were performed with ramp rates of 20 °C min−1.Results and discussion
Fig. 1 shows the powder X-ray diffractograms collected for the as-made LSM35, LSM50, LSM65 and LSM80. Fig. 1a shows that all patterns are characteristic of single phase perovskite structures. Final diagrams obtained from LeBail fits show the indexation and fitting of PXRD patterns over a 2θ range of 10 to 100° and are presented in Fig. SI1.† Fig. 1b shows that different peak profiles are clearly observed for the perovskite main reflections, highlighting the distinct crystal symmetries of the compounds, and yielding to the splitting of some reflections. Systematic single Kα doublets were only observed in the case of LSM80. Thus, in agreement with literature,30,31 distorted structures were observed for LSM35 (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c), LSM50 and LSM65 (both I4/mcm), whereas LSM80 has the ideal cubic perovskite symmetry (Pmm). These different distortions and structural evolutions are partly explained by the variations observed for the Goldschmidt tolerance factor32 that continuously evolves as the A-site and B-site ionic radii are changed in the ABO3-type LSM compounds. Fig. 1b also shows a shift of the Bragg peaks toward higher angles as Sr2+ substitutes for La3+. A decrease of the volume per formula unit is observed upon Mn3+ (r = 0.645 Å) replacement by Mn4+ (r = 0.53 Å), caused by the charge balance induced by divalent Sr2+ introduction over the trivalent La3+. Although it is not straightforward to directly compare the cell parameters along the LSM series due to the change in crystal symmetries, one can obtain a more direct comparison by establishing the structural relationships of the different distorted structures in regard of the ideal cubic perovskite. This information, gathered from the cell parameters extracted from LeBail fits is presented in Table SI1† and clearly reflects the average decrease in volume as Mn4+ substitutes for Mn3+.
c), LSM50 and LSM65 (both I4/mcm), whereas LSM80 has the ideal cubic perovskite symmetry (Pmm). These different distortions and structural evolutions are partly explained by the variations observed for the Goldschmidt tolerance factor32 that continuously evolves as the A-site and B-site ionic radii are changed in the ABO3-type LSM compounds. Fig. 1b also shows a shift of the Bragg peaks toward higher angles as Sr2+ substitutes for La3+. A decrease of the volume per formula unit is observed upon Mn3+ (r = 0.645 Å) replacement by Mn4+ (r = 0.53 Å), caused by the charge balance induced by divalent Sr2+ introduction over the trivalent La3+. Although it is not straightforward to directly compare the cell parameters along the LSM series due to the change in crystal symmetries, one can obtain a more direct comparison by establishing the structural relationships of the different distorted structures in regard of the ideal cubic perovskite. This information, gathered from the cell parameters extracted from LeBail fits is presented in Table SI1† and clearly reflects the average decrease in volume as Mn4+ substitutes for Mn3+.
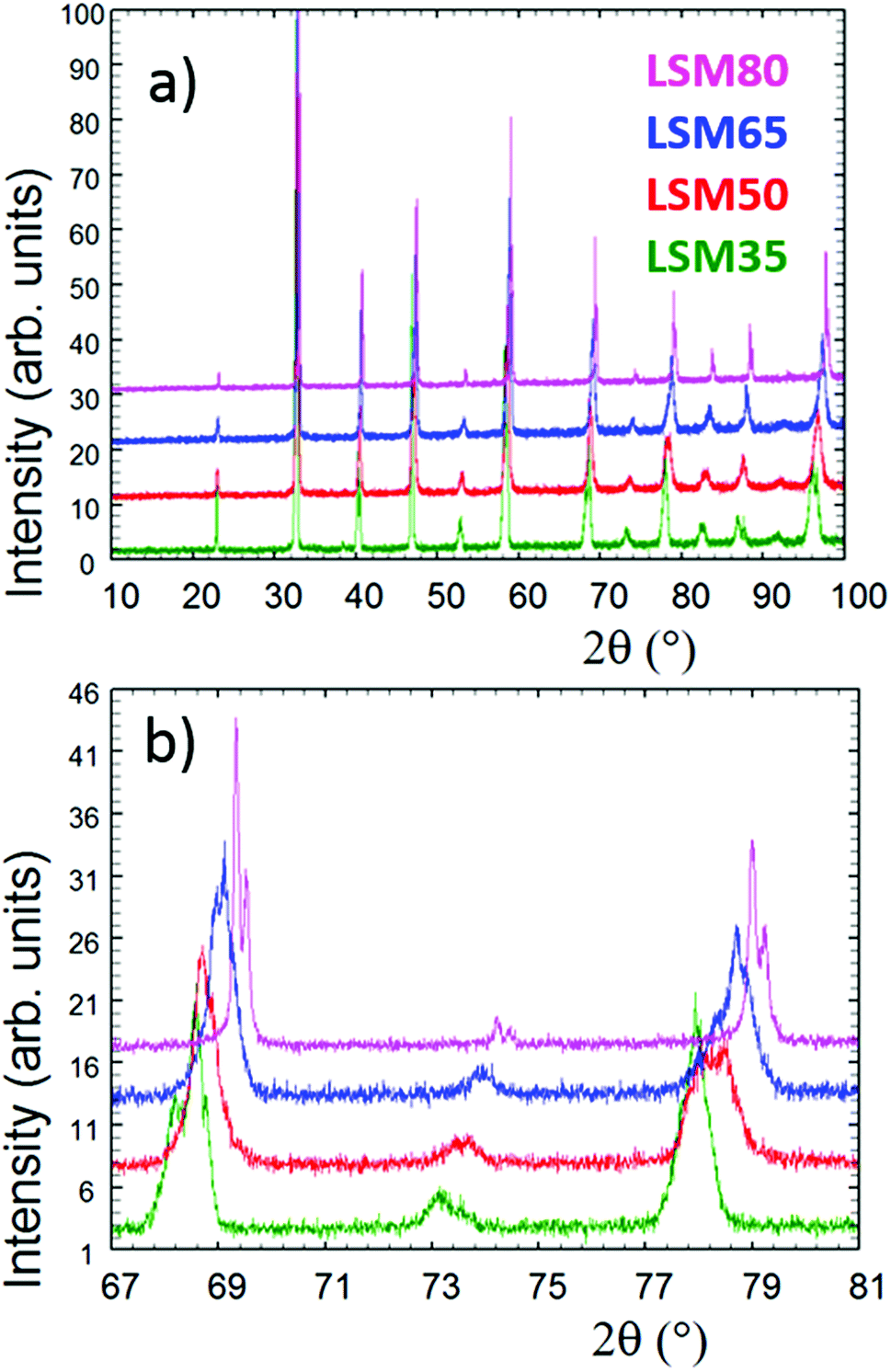 | ||
| Fig. 1 (a) Full X-ray powder diffraction diagrams collected for the as-made samples. (b) Focus on the 67–81° range showing the different peak profiles and splitting of some reflections. | ||
Thermochemical cycles were performed on the LSM series and the weight variations resulting from reduction or oxidation were collected by TG analysis. Identical thermochemical treatments were applied for all the samples, with a reduction step performed at 1400 °C for 0.75 h under Ar and a CO2 dissociation step performed at 1050 °C for 1 h. Fig. 2a focuses on the O2 production rate during the reduction whereas Fig. 2b shows two successive cycles of the full two-step thermochemical process. One should note that a slight weight drift is observed during non-isothermal heating, which can be attributed to thermal gas expansion and buoyancy effects on the crucible, and which could be considered minor in contrast to the large reduction capabilities of the samples. Within these La1−xSrxMnO3−δ series, reduction capabilities increase monotonically with the Sr content. As x increases, the O2 production rate increases, the temperature needed to initiate the reduction is lowered, and the total O2 production is enhanced. Extended to a larger x range, these results correlate well with the behavior observed for x = 0.1 to x = 0.4.27 The activation energies of the reduction reaction (details in ESI, Fig. SI2†) were determined according to a contracting sphere model,33,34 which is assumed to be the dominant representative solid-state process during partial reduction of non-stoichiometric perovskites.35 Values of 255, 142, 87 and 76 kJ mol−1 were found for LSM35, LSM50, LSM65, and LSM80, respectively, consistent with the fact that elevated Sr contents ease the reduction of the compounds within the LSM series. Since the introduction of Sr into LaMnO3 provides minor crystal structure changes, one may explain this behavior as a result of the significant increase of the Mn formal oxidation state with x, from LSM35 (Mn+3.35) to LSM80 (Mn+3.80). This significant increase of the Mn4+ content in the LSM series therefore leads to greater reduction capabilities, as Mn4+ should be reduced more favorably than Mn3+.
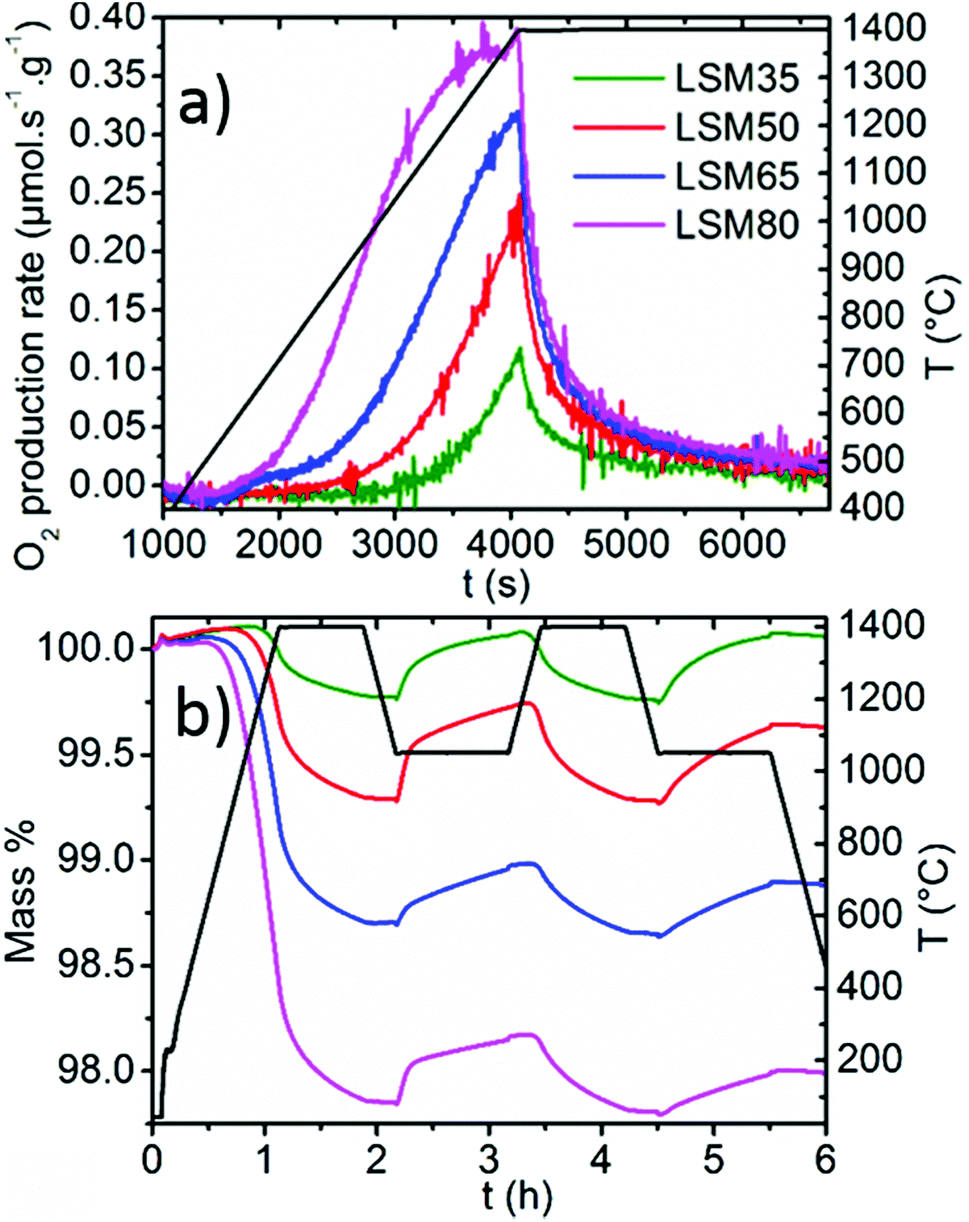 | ||
| Fig. 2 (a) O2 production rates derived from the TG data. (b) TG curves collected during reduction at 1400 °C under Ar followed by exposure to CO2 at 1050 °C (two cycles). | ||
Fig. 3a shows that the re-oxidation yield decreases steeply with x. This strongly suggests that thermodynamics for full re-oxidation are increasingly unfavorable as the Sr content is raised. LSM35 shows an almost complete re-oxidation yield with a CO production reaching close twice the O2 production, whereas the re-oxidation is significantly reduced for LSM50. For LSM65 and LSM80, only slight portions of the oxygen released by the crystal lattice during the reduction are recovered when performing the CO2 dissociation step. Accordingly, compilation of previously reported thermodynamic data clearly shows that oxidation thermodynamics are increasingly unfavorable in the La1−xSrxMnO3−δ from x = 0.2 to x = 0.4 and from x = 0 to x = 0.5.24,27 Nevertheless, elevated gravimetric productions are observed for all the compounds due to the partial re-oxidation under CO2 (Fig. 3b). Each compound of the series produces for example more than pure CeO2 for comparable thermochemical conditions,19 owing to more favorable reduction thermodynamics. LSM50 seems like the best trade-off between reduction and oxidation thermodynamics, which allows achieving CO productions of 269 and 215 μmol g−1 during the first and second cycles respectively. These values compare with those observed for the best Zr-substituted ceria compounds within similar thermochemical conditions.19 More insight on the redox behavior of the LSM series is obtained by plotting the evolution of both the oxygen non-stoichiometry and the Mn oxidation states during the two-step cycles (Fig. 4).
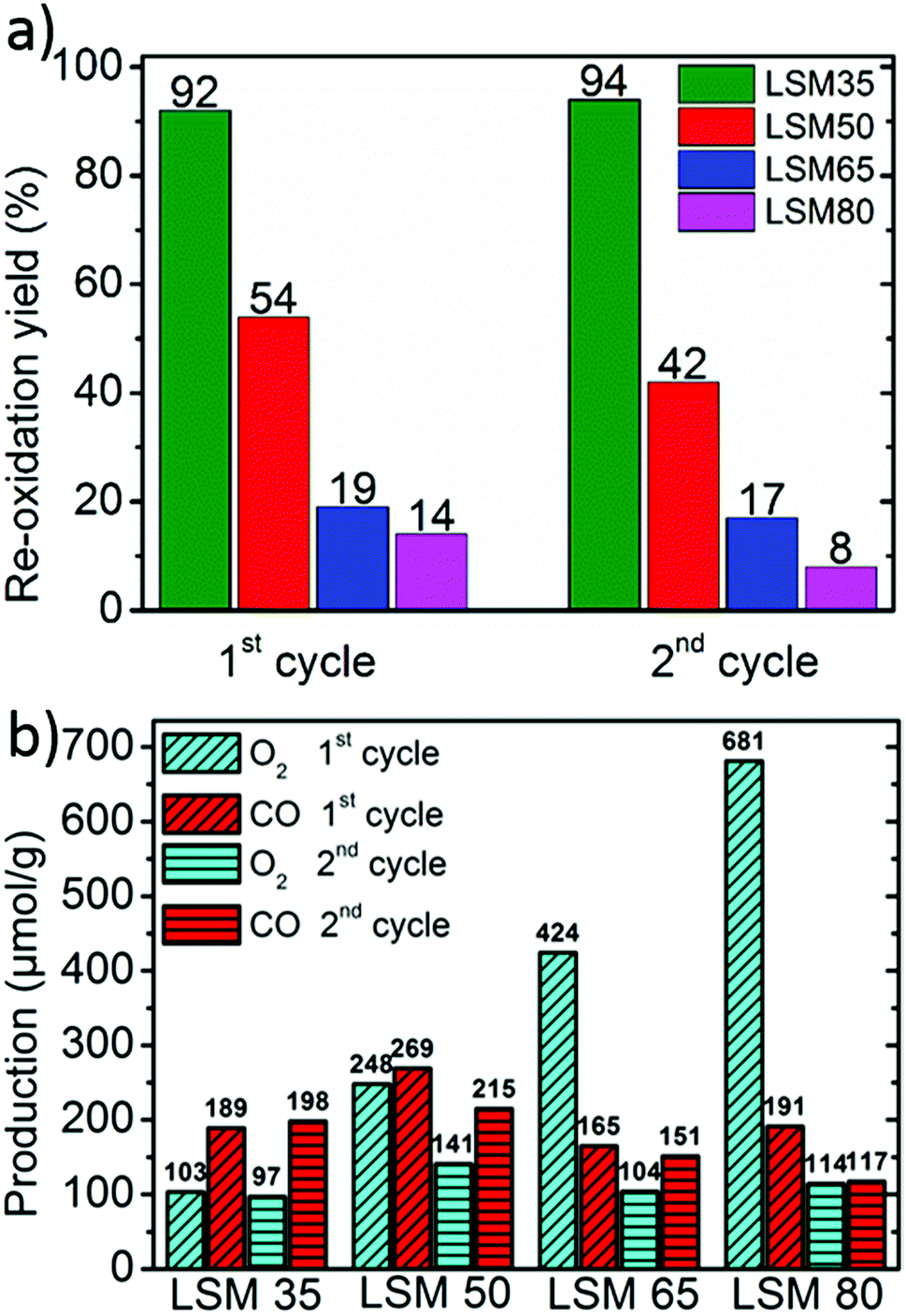 | ||
| Fig. 3 (a) Re-oxidation yields of the perovskites under CO2 exposure. (b) O2 and CO productions during the two cycles. | ||
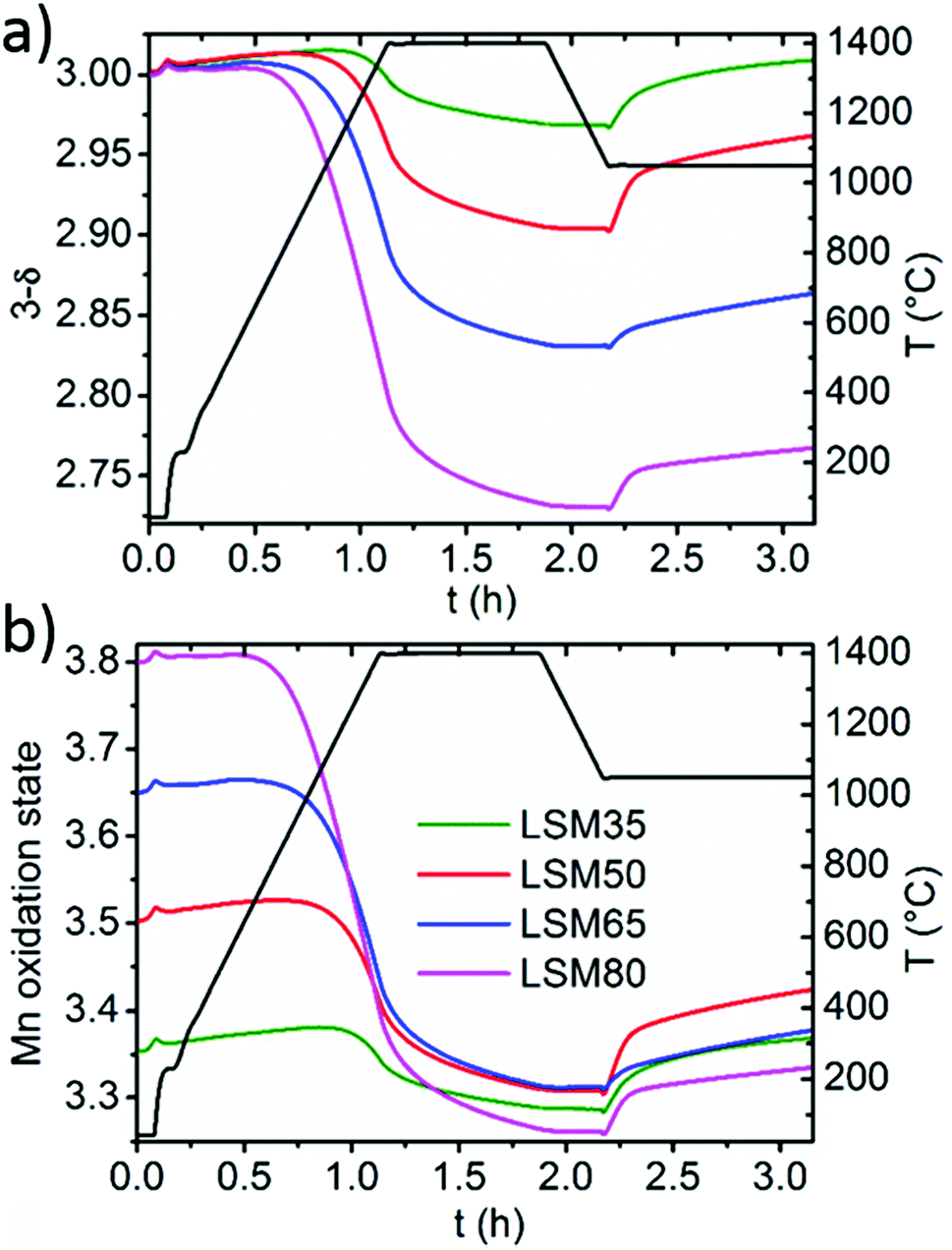 | ||
| Fig. 4 (a) Evolution of the oxygen content of the perovskites during thermal reduction under Ar (1400 °C) followed by re-oxidation under CO2 (1050 °C). (b) Evolution of the formal oxidation state of Mn under the same thermochemical treatment. | ||
Since Mn is the only redox active cation in the La1−xSrxMnO3−δ series, the evolution of its oxidation state could be calculated from the weight variations observed during the cycling experiments. Since these compounds are all containing mixed valence Mn4+/Mn3+, thermal reduction should firstly involve reduction of the Mn4+ fraction into Mn3+, and reciprocally, re-oxidation should involve reversible oxidation of Mn3+ into Mn4+. Fig. 4a illustrates well the enhancement of oxygen vacancy formation for elevated Sr contents, yielding a variety of final oxygen stoichiometries achieved after reduction at 1400 °C for 45 minutes: La0.65Sr0.35MnO2.97 (LSM35), La0.50Sr0.50MnO2.90 (LSM50), La0.35Sr0.65MnO2.83 (LSM65) and La0.20Sr0.80MnO2.73 (LSM80). Strikingly, these stoichiometries correlate with a very narrow range of reduced Mn oxidation states, regardless of the Sr content and associated oxygen release (Fig. 4b). Indeed, final reduced oxidation states of Mn are +3.29 (LSM35), +3.31 (LSM50), +3.31 (LSM65), and +3.26 (LSM80), while the initial oxidation states are +3.35, +3.50, +3.65, and +3.80 respectively. This similarity is also observed during the CO2 dissociation step, which also results in a very narrow range of final oxidation states after the re-oxidation of the LSM compounds, with extreme values of +3.33 for LSM80 and +3.42 for LSM50. Such behavior, based on a comparable redox activity of Mn for all compounds, explains why similar CO productions are achieved within the series despite a strong discrepancy in the re-oxidation yields. For example, the similar Mn oxidation states reached after both the reduction and oxidation for LSM35 and LSM80 allows them to produce respectively 189 and 191 μmol g−1, whereas the associated re-oxidation yields are 92 and 14%. Fig. 4 strongly suggests that within the range from x = 0.35 to x = 0.80, the final CO production is poorly influenced by the very distinct oxygen vacancy formation capabilities of the compounds, but it is rather governed by a similar redox activity of Mn determined by the final oxidation state obtained after reduction. The better productions observed for LSM50 therefore only correspond to a slightly larger redox activity of Mn during the thermochemical cycles, which may be related to a more favorable thermodynamic driving force when compared to LSM65 and LSM80, thereby triggering more rapid kinetics. Also, this narrow range of reactivity corresponds to a re-oxidation yield close to 100% in LSM35, which therefore explains why no larger CO production is exhibited when compared to LSM50, despite the fact that more favorable re-oxidation thermodynamics should be expected for LSM35. After the first cycle a decrease of the CO production is observed for some of the samples, as a result of powder densification and sintering that alters gas transfer to the reaction sites and surface area available for the solid–gas reaction.
Purely in term of redox activity, the LSM series of perovskites clearly shows enhanced performance when compared to fluorite type CeO2, with a larger electronic transfer during the oxidation by CO2. Indeed, while the electronic transfer involves 0.036 e− per formula unit (p.f.u.) in CeO2 under similar conditions,19 up to 0.116 e− p.f.u. are involved in the re-oxidation of LSM50. This latter value also compares favorably with the electronic transfer in Ce0.75Zr0.25O2 that reaches 0.076 e− p.f.u. (or 0.102 e− per Ce atom, which is the redox active element in this case).19
Fig. 5 shows a comparison of the re-oxidation behavior of LSM50 when reduced at 1400 °C and re-oxidized at different temperatures. Full thermochemical cycles are shown in ESI along with the corresponding re-oxidation yields (Fig. SI3†). While 1050 °C seems to be close to the optimal temperature of re-oxidation (Fig. 5a), comparable CO productions are observed during the first cycle for temperatures of 900 and 1200 °C (Fig. 5b). An alteration of the re-oxidation kinetics is also observed after the first cycle, but final CO productions remain above 200 μmol g−1 for re-oxidations performed at 1050 °C and 1200 °C. Such phenomenon is often observed during thermochemical cycling of redox active materials and corresponds to powder densification and sintering that are both unfavorable for solid–gas reactions by limiting mass transfer throughout packed powder and decreasing the available surface area for solid–gas exchange. Accordingly, the re-oxidation at 900 °C is strongly altered by this sintering, with a steep decrease of the CO production from 224 to 146 μmol g−1. Since the thermodynamic driving force for the re-oxidation should be more favorable at 900 °C than it is at 1050 or 1200 °C, one can reason this by kinetic limitations. In particular, a larger influence of the diffusion regime is to be expected after sintering of the particles. Since oxide ion bulk diffusion is thermally activated, sintering of the particles should have a larger impact on the kinetics at lower temperatures. Re-oxidation at 1400 °C (isothermal cycling) is shown to be highly unfavorable when compared to lower temperatures, which agrees well with the fact that better performances are to be achieved with re-oxidation temperatures lower than reduction temperatures for thermodynamic reasons. However, it should also be noted that to our knowledge, the CO production of 108 μmol g−1 achieved with LSM50 at 1400 °C represents the largest production ever reached by two-step isothermal CO2 splitting with an oxide material.
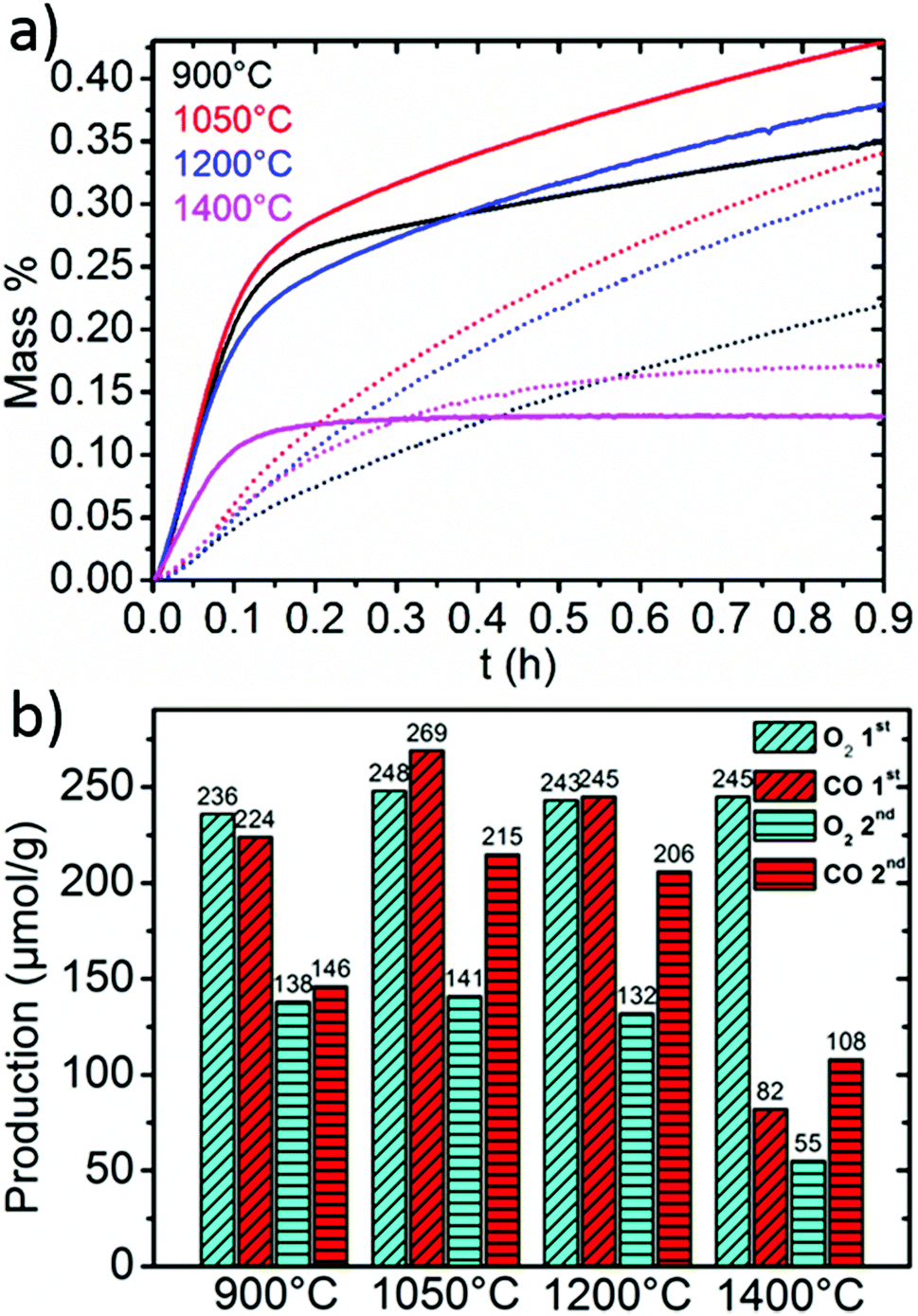 | ||
| Fig. 5 (a) Mass uptake evolution for LSM50 during re-oxidation at 900, 1050, 1200 and 1400 °C, after thermal reduction performed at 1400 °C. Lines and dots refer to the first and second re-oxidation respectively. (b) O2 and CO productions during the two cycles. | ||
In addition, three successive cycles were performed at the re-oxidation temperature of 1050 °C for LSM50 (Fig. SI4†). Fig. 6 focuses on the re-oxidation steps to characterize the thermal stability of the material, with repeatable CO productions and kinetics observed after the second cycle. Indeed, the alteration of performances observed after the first cycle does not occur in the subsequent cycles. Strikingly, the re-oxidation curves for the second and third cycles are almost identical, suggesting that the effect of sintering could be negligible after the first cycle. Moreover, the fact that all La1−xSrxMnO3−δ compounds examined here have been synthesized and stabilized at temperatures larger than 1400 °C (which is the maximal temperature employed here in the thermochemical cycles) during several sequences of grinding and firing strongly suggests that these perovskites should offer long term thermal and chemical performance stability at the temperature required for the thermochemical cycles used here. The non-stoichiometric behavior should also be seen as a favorable parameter for this matter, conferring flexibility to these compounds and allowing them to be stable in a wide range of temperature and oxygen partial pressure.36–39
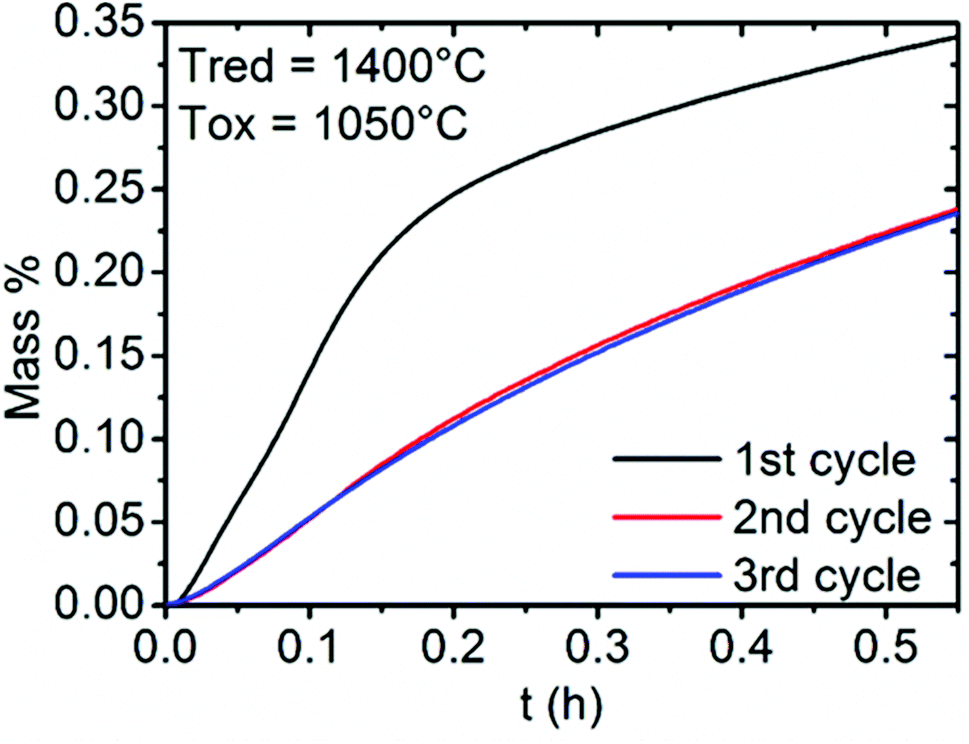 | ||
| Fig. 6 Evolution of the reactivity of LSM50 during three successive thermochemical cycles. Only the mass uptakes during the re-oxidation steps by CO2 are shown. | ||
Conclusions
Thermochemical CO2 splitting via redox cycling of LSM compounds generates CO productions that are comparable with state of the art materials such as CeO2, and that are associated with a high redox activity of the Mn4+/Mn3+ couple toward CO2 dissociation, resulting in larger electronic transfers than those observed in ceria during the re-oxidation step. For this particular series of compounds, this implies that if the superior redox activity of Mn in these perovskites can be retained through substitution, incorporation of lighter elements such as Ca2+ for Sr2+ or Y3+ for La3+ maybe beneficial to the final CO productions. More generally, the vast possibilities offered by chemical tuning with multiple substitutions allowed in perovskites, combined with an optimization of the microstructure to favor the solid–gas reactions, appear as very promising in order to conceive efficient redox catalysts for solar two-step thermochemical dissociation of CO2.To a certain extent, the controlled introduction of Sr into LaMnO3 allows tuning the redox thermodynamics in the La1−xSrxMnO3 series. For instance, our results strongly correlate with a monotonic decrease of the free enthalpy of reduction with x. As such, the La1−xSrxMnO3 series is a good example of system for which the two end members (x = 0 and x = 1) display distinct enough redox properties, so that the intermediate compounds can be suitable for elevated activity toward thermal reduction followed by CO2 dissociation. This pathway to generate efficient redox catalysts for two-step thermochemical processes should be available in numerous solid solutions of mixed valence perovskites.
Interestingly, the Mn4+/Mn3+ redox couple is predicted to be thermodynamically inert toward CO2 splitting within stoichiometric oxides such as the MnO2/Mn2O3 redox pair. Even the Mn3+/Mn2+ is still inactive in simple manganese oxides (Mn3O4/MnO redox pair being impractical in two-step CO2 splitting cycles). In contrast, for all compounds examined here, the evolution of the Mn oxidation state implies partial re-oxidation of Mn3+ into Mn4+, and the CO2 dissociation step therefore involves the Mn4+/Mn3+ redox pair. The fact that this couple can be activated within the LSM series is both a promising and intriguing feature of this family of materials, showing that, in the context of thermochemical CO2 dissociation, non-stoichiometric perovskites could open access to redox couples that would conventionally not be considered with simple stoichiometric oxides.
Acknowledgements
This study was funded by Airbus Group Corporate Foundation (CNRS CT 084210) and by CNRS (défi ENRS, VALTHER-CO2 project). The authors also thank D. Perarnau and E. Bèche for collection of XRD data and fruitful discussions.Notes and references
- A. Steinfeld, Sol. Energy, 2005, 78, 603–615 CrossRef CAS PubMed.
- T. Kodama and N. Gokon, Chem. Rev., 2007, 107, 4048–4077 CrossRef CAS PubMed.
- J. E. Miller, M. D. Allendorf, R. B. Diver, L. R. Evans, N. P. Siegel and J. N. Stuecker, J. Mater. Sci., 2008, 43, 4714–4728 CrossRef CAS PubMed.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed.
- T. Nakamura, Sol. Energy, 1977, 19, 467–475 CrossRef CAS.
- Y. Tamaura, A. Steinfeld, P. Kuhn and K. Ehrensberger, Energy, 1995, 20, 325–330 CrossRef CAS.
- Y. Tamaura, M. Kojima, T. Sano, Y. Ueda, N. Hasegawa and M. Tsuji, Int. J. Hydrogen Energy, 1998, 23, 1185–1191 CrossRef CAS.
- S. B. Han, T. B. Kang, O. S. Joo and K. D. Jung, Sol. Energy, 2007, 81, 623–628 CrossRef CAS PubMed.
- H. Kaneko, T. Yokoyama, A. Fuse, N. Ishihara and Y. Tamaura, Int. J. Hydrogen Energy, 2006, 31, 2256–2265 CrossRef CAS PubMed.
- T. Kodama, N. Gokon and R. Yamamoto, Sol. Energy, 2008, 82, 73–79 CrossRef CAS PubMed.
- N. Gokon, H. Muramyama, A. Nagasaki and T. Kodama, Sol. Energy, 2009, 83, 527–537 CrossRef CAS PubMed.
- F. Fresno, R. Fernandez-Saavedra, M. B. Gomez-Mancebo, A. Vidal, M. Sanchez, M. I. Rucandio, A. J. Quejido and M. Romero, Int. J. Hydrogen Energy, 2009, 34, 2918–2924 CrossRef CAS PubMed.
- F. Fresno, T. Yoshida, N. Gokon, R. Fernandez-Saavedra and T. Kodama, Int. J. Hydrogen Energy, 2010, 35, 8503–8510 CrossRef CAS PubMed.
- S. Abanades, A. Legal, A. Cordier, G. Peraudeau, G. Flamant and A. Julbe, J. Mater. Sci., 2010, 45, 4163–4173 CrossRef CAS.
- W. C. Chueh and S. M. Haile, ChemSusChem, 2009, 2, 735–739 CrossRef CAS PubMed.
- M. Kang, X. M. Wu, J. Zhang, N. Zhao, W. Wei and Y. H. Sun, RSC Adv., 2014, 4, 5583–5590 RSC.
- J. R. Scheffe, R. Jacot, G. R. Patzke and A. Steinfeld, J. Phys. Chem. C, 2013, 117, 24104–24114 CAS.
- A. Legal and S. Abanades, Int. J. Hydrogen Energy, 2011, 36, 4739–4748 CrossRef CAS PubMed.
- A. Legal, S. Abanades and G. Flamant, Energy Fuels, 2011, 25, 4836–4845 CrossRef CAS.
- A. Legal and S. Abanades, J. Phys. Chem. C, 2012, 116, 13516–13523 CAS.
- A. Evdou, L. Nalbandian and V. T. Zaspalis, J. Membr. Sci., 2008, 325, 704–711 CrossRef CAS PubMed.
- A. Evdou, V. Zaspalis and L. Nalbandian, Int. J. Hydrogen Energy, 2008, 33, 5554–5562 CrossRef CAS PubMed.
- L. Nalbandian, A. Evdou and V. Zaspalis, Int. J. Hydrogen Energy, 2009, 34, 7162–7172 CrossRef CAS PubMed.
- J. R. Scheffe, D. Weibel and A. Steinfeld, Energy Fuels, 2013, 27, 4250–4257 CrossRef CAS.
- A. H. McDaniel, E. C. Miller, D. Arifin, A. Ambrosini, E. N. Coker, R. O'Hayre, W. C. Chueh and J. H. Tong, Energy Environ. Sci., 2013, 6, 2424–2428 CAS.
- A. Demont, S. Abanades and E. Beche, J. Phys. Chem. C, 2014, 118, 12682–12692 CAS.
- C.-K. Yang, Y. Yamazaki, A. Aydin and S. M. Haile, J. Mater. Chem. A, 2014, 2, 13612–13623 CAS.
- J. Rodriguez-Carvajal, Fullprof, http://www.ill.eu/sites/fullprof/php/programs.html Search PubMed.
- T. Roisnel and J. Rodriguez-Carvajal, Mater. Sci. Forum, 2001, 118, 278–381 Search PubMed.
- P. M. Woodward, T. Vogt, D. E. Cox, A. Arulraj, C. N. R. Rao, P. Karen and A. K. Cheetham, Chem. Mater., 1998, 10, 3652–3665 CrossRef CAS.
- O. Chmaissem, B. Dabrowski, S. Kolesnik, J. Mais and J. D. Jorgensen, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 094431 CrossRef.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 21, 477–485 CrossRef.
- F. J. Gotor, J. M. Criado, J. Malek and N. Koga, J. Phys. Chem. A, 2000, 104, 10777–10782 CrossRef CAS.
- A. Khawam and D. R. Flanagan, J. Phys. Chem. B, 2006, 110, 17315–17328 CrossRef CAS PubMed.
- M. A. Pena and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2017 CrossRef CAS PubMed.
- J. Mizusaki, N. Mori, H. Takai, Y. Yonemura, H. Minamiue, H. Tagawa, M. Dokiya, H. Inaba, K. Naraya, T. Sasamoto and T. Hashimoto, Solid State Ionics, 2000, 129, 163–177 CrossRef CAS.
- J. Mizusaki, Y. Yonemura, H. Kamata, K. Ohyama, N. Mori, H. Takai, H. Tagawa, M. Dokiya, K. Naraya, T. Sasamoto, H. Inaba and T. Hashimoto, Solid State Ionics, 2000, 132, 167–180 CrossRef CAS.
- J. H. Kuo, H. U. Anderson and D. M. Sparlin, J. Solid State Chem., 1989, 83, 52–60 CrossRef CAS.
- M. Andrieux and C. Picard, J. Mater. Sci. Lett., 2000, 19, 695–697 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: LeBail fits of PXRD diagrams and further thermochemical characterizations (TGA). See DOI: 10.1039/c4ra10578h |
| This journal is © The Royal Society of Chemistry 2014 |